В главе «Методы» я писал о том, как наша группа – З.А. Авенирова, Н.В. Энгельгардт и я пришли к иммунодиффузии, к чему вскоре присоединились А.И. Гусев и В.С. Цветков, и как мы начали иммунодиффузионный анализ фракции цитоплазматических гранул печени – митохондрий и микросом, получаемых на сепараторе.
Вначале с полной достоверностью был обнаружен органоспецифический антиген печени (А0) и несколько других антигенов этой группы (см. раздел об органоспецифических антигенах), затем – в виде слабой тени – антиген, специфичный для гепатомы (АГ) (Зильбер и др. 1959). Все наши первоначальные опыты были проведены с мышиной гепатомой XXIIa, полученной Витой Ильиничной Гельштейн в 1951 г. индукцией ортоаминоазо-толуолом. В дальнейшей работе были использованы и другие гепатомы, полученные В.И. Гельштейн. Под сильным давлением Л.А. Зильбера мы сконцентрировались на АГ и вскоре, применив вместо фракции митохондрий и микросом (Мм) простой экстракт гепатомы и истощив антисыворотку к Мм экстрактами нормальных тканей, мы смогли получить сначала моноспецифическую сыворотку к АГ, а затем элюат антител к нему, четко выявлявший индивидуальный антиген в экстракте гепатомы, при его отсутствии в экстрактах печени, селезенки и почки нормальных мышей (Абелев и Авенирова, 1960). Именно для выделения АГ был разработан метод иммунофильтрации, а АГ, обладавший высокой электрофоретической подвижностью α-глобулина, был как бы специально предназначен для очистки этим методом (см. «Методы»). Вскоре АГ был очищен (Абелев и Цветков, 1960; Abelev & Tsvetkov, 1961), и мы были первыми обладателями очищенного «специфического опухолевого антигена», хотя множество сомнений и одолевало нас. Среди них главные – возможность загрязнения нашей перевиваемой гепатомы посторонним вирусом-пассажиром, либо возникновение нашей гепатомы не из гепатоцитов, а из холангиоцитов (1), имеющих свой тканеспецифический антиген, сохраняющийся в опухоли. Естественно было также ожидать, что АГ просто гиперэкспрессирован в гепатоме, но в малых количествах присутствует в нормальной печени. Проверкой этих сомнений мы и занимались в 1961 г., когда направление работы резко и неожиданно изменилось (см. Abelev 1983 г., 1989 г.). В процессе других исследований – по онтогенетическому становлению спектра органоспецифических антигенов печени – в качестве то ли дополнительного контроля, то ли без всякой «задней мысли» мы взяли также и тест-систему на АГ. Работу эту начали Н.И. Храмкова (Куприна) и студентка, которая только приступила к стажировке в лаборатории. Тест-системы на органоспецифические антигены и на АГ были «заряжены» с экстрактом эмбриональной печени, как обычно, на ночь, а результаты должны были быть назавтра утром. Студентка на следующий день не пришла, а когда Неля Храмкова посмотрела ее результаты и показала их мне – мы были ошеломлены. Нигде и никогда мы не встречали столь большой концентрации АГ – у нас просто не было материала, который мог бы дать такой артефакт. Немедленная проверка подтвердила результат – гепатомный антиген в огромных количествах был в эмбриональной печени. Проверили другие органы эмбриона – опять АГ в очень больших концентрациях. Проверили сыворотку крови – титр АГ до 1/2000 – 1/4000, в сотни раз больше, чем в гепатоме, и много больше, чем в экстрактах из органов эмбриона – значит туда он, скорее всего, попадает вместе с кровью. Проверили сыворотки мышей с гепатомой – уровень АГ в сыворотке носителей опухолей существенно выше, чем в опухоли. Значит, АГ попадает в опухоль тоже с кровью, которая всегда в экстракте опухоли присутствует, несмотря на тщательное отмывание. Но откуда он попадает в кровь – из опухоли или, может быть, из печени, как белок острой фазы, вырабатываемый в ответ на рост опухоли? Эта печальная возможность казалась нам наиболее вероятной, тем более, что незадолго до того появилась серия работ англичанина Darcy, показавшего, что в крови крыс при росте различных сарком появляется некий α-глобулин, наподобие С-реактивного белка при инфекциях. Такой белок с кровью мог попасть в гепатому и симулировать специфический для нее антиген, тем более, что в качестве контроля мы брали печень, органы и кровь нормальных, здоровых животных, без каких-либо опухолей. Мы сразу же нашли в лаборатории мышей, привитых саркомами и взяли их кровь для проверки на АГ. У одной в крови оказался АГ! Круг был замкнут – наихудшее предположение подтвердилось. Оставалась лишь одна маленькая надежда. Может быть, эта мышь была беременна, и в кровь ее попал АГ из сыворотки эмбрионов? В клетке, где сидели эти мыши, действительно были только что родившиеся мышата, но у той ли мыши они родились?
Мы тут же отыскали самцов, привитых канцерогенными саркомами и другими опухолями, и проверили их на АГ. Антиген у них не обнаруживался – это давало некоторую надежду, но стало ясно, что без обнаружения места синтеза АГ – опухоль или печень? – наши результаты по гепатомному антигену висят в воздухе. Надо было исключить организм мыши из системы – и мы вместе с З.А. Постниковой, С.Д. Перовой (2) и И.С. Ирлиным повели работу по трем направлениям – гетеротрансплантации мышиной гепатомы крысам, обработанным кортизоном, перевивке клеток гепатомы хомякам в защечный мешок и культивированию гепатомы in vitro. Не буду говорить, как мы ждали результатов этих опытов и с каким настороженным скептицизмом к ним относились . Первыми вышли опыты с хомяками, – маленькие опухолевые узелки в их защечных мешках содержали АГ. Но, может быть, это был занос антигена с привитой опухолью? Где доказательства, что АГ синтезировался трансплантированной гепатомой?
Затем – гетеротрансплантация крысам. В крови у них появлялся АГ, причем динамика его роста и падения соответствовала быстрому развитию гепатомы и ее столь же быстрому рассасыванию. Но, как известно, пуганая ворона куста боится. Может быть, у крыс есть свой АГ, близкий по антигенным свойствам мышиному (как сывороточные альбумины), и крыса отвечает его образованием на рост гетеротрансплантата? Проверили сыворотку крысиного эмбриона с тест-системой на наш АГ. Полоса преципитации с крысиной сывороткой не формировалась, но полоса нашей тест-системы «съедалась» эмбриональной сывороткой крысы. Стало ясно, что крысы имеют свой эмбриональный антиген, родственный, но не идентичный АГ, и выявляемый реакцией подавления преципитации. Может быть, крысиный антиген симулировал появление АГ при гетеротрансплантации? Так что и эти опыты нельзя было признать полностью убедительными.
Положение еще усугублялось тем, что в июле того же, 1962 г., в Москве должен был состояться VIII Международный противораковый конгресс – большое государственное событие, к которому весьма торжественно готовились. Петр Николаевич Грабар, директор Государственного института рака в Вильжуифе (Париж), организовывал специальную дискуссию по иммунологии рака на конгрессе. Он был в Москве на Всемирном биохимическом конгрессе в 1961 г., знал наши работы по АГ, доложенные на конгрессе (Абелев и др. 1961) и пригласил меня участвовать с докладом на этой дискуссии. Тезисы были посланы ему в 1961 г., когда мы еще не знали об эмбриональном происхождении АГ, и нам надо было сказать на съезде, что же с ним произошло. Времени было совсем мало, в Институте готовились к приему гостей с конгресса, в наших комнатах шел ремонт – всюду были груды мусора, работали штукатуры и маляры. Мы работали, ничего не видя вокруг.
Наконец, опыты по культурам дали результат – АГ накапливался в культуральной среде! Круг был снова замкнут! Но уже совсем по-другому. Стало ясно, что АГ, который мы переименовали уже в αF (эмбриональный – fetal – сывороточный α-глобулин), синтезируется, скорее всего, в печени эмбриона и секретируется ею в кровь. После рождения, в течение месяца уровень αF в крови падает ~ в 2000 раз, до подпороговой величины. При возникновении в печени гепатомы его синтез возобновляется, и он снова секретируется в кровь. С кровью он попадает в различные органы животного. Стало ясно, что аналогичная ситуация имеет место и у крыс – перекрестно реагирующий с αF антиген обнаруживался не только в крови крысиных эмбрионов, но и у взрослых крыс с гепатомой Зайдела.
Этот цикл работ был начат в феврале и закончен в мае 1962 г., а 24 июля я доложил эти данные на конгрессе, на заседании, которое вел П.Н. Грабар. Результаты были услышаны, причем, не только на конгрессе, но и при посещении отдела и лаборатории всеми иммуно-онкологами, бывшими на конгрессе, – Болдуином (R. Bаldwine, Англия), Шогреном (H.О. Sjögren), Хельстромами (К.Е. & I. Hellströms, Швеция), Слеттенмарк (В. Slettenmark, Швеция), Рейфом (A. Reif, США), Бюртэном (P. Burtin, Франция) и др. Кроме того, перед конгрессом с отделом и лабораторией знакомился Честер Саутем (Ch. Southam, США) из Института Слоан-Кеттеринга в Нью-Йорке. Он помогал мне переводить конгрессный доклад. Конгресс проходил в Кремлевском дворце съездов и в МГУ. Его работа широко освещалась в общей и научной прессе. Наши данные излагались в обзорах по работе секций (Абелев и Ирлин, 1963). В январско-февральском номере 1963 г. Acta Unio Contra Cancrum, содержащем труды конгресса, полный текст доклада вышел с указанием даты конгресса, 22.VII – 28.VII 1962 г. (Abelev, 1963), а в сентябре 1963 г. доклад вышел в трудах конгресса (Абелев, 1963). Сразу же после конгресса состоялся Советско-Французский симпозиум по биологии опухолей, где эти данные докладывались уже в более широком контексте антигенной структуры печени и гепатом, но обсуждались, главным образом, результаты по αF (Абелев и др., 1963; Abelev et al., 1964). В августе 1962 г. мы направили две развернутые статьи – в «Биохимию» (Абелев и др. 1963) и в «Transplantation» (Abelev et al. 1963) (3). В Transplantation – потому, что главный редактор журнала L. Brent обратился к Л.А.Зильберу с предложением прислать статью для нового журнала. Л.А. предложил мне послать нашу работу, и мы послали статью по синтезу эмбрионального сывороточного α-глобулина перевиваемой (transplantable) гепатомой мышей. Статья вышла во втором номере нового журнала в феврале 1963 г. К этому времени мы уже знали, что αF синтезируется у мышей с регенерирующей печенью (но не у крыс). Естественно, что нашей первой гипотезой о причинах реэкспрессии αF в гепатомах было сопряжение этого процесса с пролиферацией гепатоцита, регуляция которой одновременно распространялась и на продукцию αF. Это предположение и было высказано в наших первых статьях в «Биохимии» и «Transplantation». Л.А.Зильбер, который высоко оценил эти результаты, хотел, при этом, чтобы мы весь свой потенциал направили на идентификацию и выделение собственно гепатомного антигена, слабый намек на который обнаруживался в наших опытах. Но нам уже невозможно было оторваться от αF. Мы продолжали активную работу в этом направлении. Были получены антисыворотки к эмбриональной сыворотке мышей и крыс, которые после истощения нормальной сывороткой взрослых животных оставляли антитела у мышей к αF, а у крыс – к двум четким антигенам эмбриональной сыворотки – одному с подвижностью α1-глобулина, почти идентичной подвижности альбумина, и другому – с подвижностью α2. α1 был крысиным аналогом αF, т.к. давал с ним четкую реакцию частичной идентичности, имея очень высокий титр порядка 1/2000. Он не индуцировался ни частичной гепатэктомией, ни воспалением, вызванным скипидаром у взрослых животных и появлялся в крови только при гепатоме Зайдела. α2, наоборот, имел невысокий уровень в крови новорожденных крысят, не давал перекрестных реакций с мышиным αF и четко индуцировался обработкой кожи скипидаром или при частичной гепатэктомии. Это явно был белок острой фазы. Оба антигена были описаны нами в 1965 г. в статье, которая, к сожалению, пролежала в «Вопр. мед. химии» до 1967 г. (Перова и Абелев, 1967). К 1964 г. мы знали об αF уже довольно много: его появление специфично для разных гепатом, хотя не для всех – первичные гепатомы его либо не продуцируют вовсе, либо в очень малых количествах. Связь продукции αF со степенью дифференцировки и корреляция с пролиферацией гепатоцитов были очевидными.
Итак, наша первая гипотеза – пролиферация. Но долго она не продержалась. Во-первых, регенерация печени у крыс протекала без заметной продукции αF. Гепатэктомия до 2/3 печени у крыс так же, как и у мышей, ведет к синхронной волне делений гепатоцитов, быстро восстанавливающей печень. Причем у мышей регенерация печени сопровождается невысокой, но явной волной αF, а у крыс αF при регенерации печени вообще не обнаруживался. То же при отравлении печени CCl4 у мышей. В этих случаях была очевидна большая разница (~ десятикратная) в динамике αF при сходстве, если не идентичности волн пролиферации гепатоцитов. Правда, у очень молодых крыс (5 недель), у которых только что исчез из крови αF, гепатэктомия вела к синтезу αF. Много позже, при резком увеличении чувствительности метода определения αF мы обнаружили его при регенерации печени у взрослых крыс (Перова и др. 1971). Но это только подтверждало разницу с мышами – у взрослых крыс и мышей пролиферация одинакова, а динамика αF – резко различна. Мы нашли работу в литературе, показывающую, что у крыс в ранний постнатальный период имеется 10-дневный перерыв в пролиферации клеток печени. Однако, никакого провала в уровне αF в первые дни постнатального периода крыс не наблюдалось. Мы, вместе с Ринадом Бакировым, считали митозы в постнатальной печени крыс и смотрели параллельное включение С14-аминокислот в αF. Никакой корреляции между индексом пролиферации и включением метки мы не нашли (Abelev & Bakirov, 1967). При этом связь продукции αF со степенью дифференцировки явно выявлялась в гепатомах и в онтогенезе печени (Abelev, 1965), но как тогда объяснить синтез αF при регенерации взрослой печени у мышей? Там ведь пролиферировали зрелые дифференцированные гепатоциты. В дальнейшем, при иммуногистохимическом изучении действия CCl4 у мышей мы очень четко показали, что в одной и той же дольке печени гепатоциты, расположенные вокруг некроза и в глубине от него, не различаясь по отношению к пролиферативному пулу, были прямо противоположны по синтезу в αF (см. ниже).
Наша следующая гипотеза исходила из аналогии между синтезом αF и эмбрионального и фетального гемоглобинов (Hbε и HbF). Смена эмбрионального Hbε на фетальный (HbF) и фетального на взрослый происходит (или, скорее всего, происходит) за счет смены разных популяций эритроцитов, которые развиваются из эмбриональной и фетальной стволовой клетки. В онтогенезе происходит замена ветвей дифференцировки – эмбриональной на фетальную, фетальной – на взрослую. Таким образом, идет созревание эритропоэза. Эритробластный лейкоз, скорее всего, возникает путем злокачественной трансформации эритробласта, имеющего соответствующую детерминацию, а не путем дедифференцировки более зрелой формы.
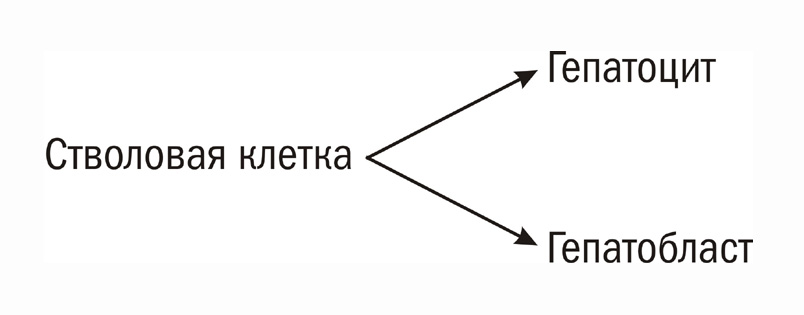
Такая схема эритроидной дифференцировки не требовала ни трансдифференцировки, ни дедифференцировки при возникновении эритробластоза эмбрионального или фетального типа. А нам остро не хотелось ни того, ни другого – ни транс- ни де-дифференцировки. Дифференцировку мы предпочитали рассматривать как необратимый процесс. В то же время Уриель легко принимал и одно и другое и рассматривал αF как следствие ретродифференцировки печени (Uriel, 1971) (4). Мы предположили, что существует стволовая клетка печени, дающая две ветви дифференцировки – эмбриональную (гепатобласты) и взрослую (гепатоциты). αF является маркером эмбриональной ветви. При регенерации может возникать (мыши) или не возникать (крысы, человек) эмбриональная ветвь, соответственно, и в αF. Гепатомы, возникающие из стволовой клетки или ее ближайшего потомства, сохраняют способность к альтернативной дифференцировке, в которой преобладает либо одна, либо другая ветвь, либо обе отсутствуют (Abelev, 1968). Эта гипотеза нам нравилась своим биологизмом, а также тем, что не требовала дедифференцировки. Она была также весьма конструктивной и имела конкретные экспериментальные следствия. Гипотеза предсказывала, что:
а) смена αF на взрослый тип сывороточных белков происходит не путем затухания синтеза αF, а путем смены клеточной популяции: αF-положительной на αF-отрицательную;
б) возобновление синтеза αF при регенерации печени у мышей должно сопровождаться появлением его в отдельных клетках, а не возобновлением синтеза во всех гепатоцитах;
в) разница в продукции αF различными гепатомами обусловлена соотношением «αF+» и «αF-» популяций, возникающих в опухоли из трансформированной стволовой клетки печени;
г) в печени, весьма вероятно, существуют отдельные, моноклональные популяции гепатоцитов, синтезирующих αF или альбумин;
д) популяция αF-продуцирующих клеток соответствует конечной стадии эмбриональной ветви дифференцировки и представляет собой клетки, выходящие из пролиферативного пула злокачественных клеток.
Предсказаний было много – вполне конкретных и экспериментально проверяемых, что от гипотезы и требовалось.
Прежде всего, необходима была ИФ локализация αF, но она упорно не удавалась (см. «Методы»). Наконец, применение метода Сэнт-Мэри для фиксации и окраски печеночных клеток на сывороточный альбумин позволило увидеть αF в эмбриональной печени (Энгельгардт и др., 1969).
На Лондонской конференции Chester-Вeatty Института по раку печени английский патолог Edington, работавший в Африке по раку печени, очень ворчливо и недоверчиво заявил: «Абелев сказал, что у них есть метод локализации αF-глобулина на срезах – пусть он предъявит нам методику». Я был очень рад этому требованию, у меня было уже с собой, сделанное Н.В. Энгельгардт, описание техники локализации αF. Я его (уже не помню как) размножил и распространил на конференции.
На той же конференции мы крепко столкнулись с Уриелем по поводу αF у мышей и крыс. Я уже писал, что в январе 1965 г. после конференции в Монте-Карло, П.Н. Грабар пригласил Л.А. Зильбера и меня посетить его лабораторию в Институте рака в Вильжуифе и в Пастеровском Институте в Париже. Тогда я познакомился с Уриелем, Станиславским-Биренцвайгом, Аврамисом, Бюртеном и Сабиной фон Клейст. Станиславский и Уриель показали мне свои данные по крысам, которые были один к одному с нашими, о чем я и сказал, увидев иммунофореграммы Станиславского и Уриеля. Единство было полное. Станиславский и Уриель опубликовали свои результаты в Cancer Res. в 1967 г., мы – в Вопр. Мед. Химии (Перова и Абелев, 1967) после 2-х летнего лежания статьи в редакции, о чем я уже писал. И хотя Уриель знал о наших результатах, но в предварительном тексте доклада к Лондонской конференции он писал, что мы обнаружили у мышей антиген, соответствующий то ли эмбриональному α1, то ли α2 – белку острой фазы. Я удивился и обиделся и написал ему письмо с сожалением, что мнение столь авторитетной лаборатории и ученого вносит путаницу в ясный вопрос. Мы со Светой Перовой специально подготовили демонстрацию, в которой эмбриональные сыворотки мышей и крыс подвергались электрофорезу друг за другом и проявлялись соответствующими антисыворотками. При этом αF мышей и крыс легко идентифицировались и давали реакцию частичной идентичности.
Картина была совершенно однозначная, указывающая на наличие родственных αF у мышей и крыс. Эту демонстрацию я показал на конференции в дискуссии после доклада Уриеля. Больше у нас недоразумений на эту тему не возникало и в статье Уриеля, опубликованной по материалам конференции, этот вопрос был представлен правильно.
В общем конференция была очень интересной и полезной. Там я познакомился с канадцем Фарбером (Farber), открывшим овальные клетки в печени, биохимиком печени – голландцем Эммелотом (Emmelot), Парвесом (Purves) из ЮАР, работавшим по αF в Мозамбике и ЮАР с РИА. Ему я дал нашу анти- αF-человека и рассказал об иммунизации в лимфоузел, чему он был и рад и удивлен и потом нас сердечно благодарил за иммунизацию в лимфоузел, сообщенную ему еще до публикации.
Кстати, я гостил тогда и у Maurine Dale, нашей африканки-англичанки и у ее друзей. Но это уже отдельный сюжет. Возвращаясь к гипотезе о стволовой клетке печени и о двух субпопуляциях гепатоцитов, первое, что необходимо было сделать – это выяснить, как на клеточном уровне происходит угасание продукции αF в постнатальной печени мышей и крыс. И мы начали эту работу с Люсей Шиповой, когда Наташа Энгельгардт была в Африке. Работа четко показала, что угасание в αF идет путем смены популяций «αF+» на «αF-». Но смена эта была своеобразной: популяция «αF+» клеток угасала градиентно, она как бы «сжималась» вокруг сосудов (центральных вен) (Шипова и др. 1974) – феномен, впоследствии многократно подтвержденный. Такое затухание, когда по периферии градиента αF угасал, а не замещался, говорило и «за» и «против» гипотезы замещения. Гипотеза оставалась неопределенной, но сам факт «градиентного затухания» синтеза αF в постнатальной печени стал одним из основополагающих в клеточном анализе αF. Он был описан также через 25 лет на молекулярном уровне (Spear, 1999) (5).
Другая линия клеточного исследования αF – его локализация в гепатомах. Наташа Энгельгардт и Толя Гусев были во время Лондонской конференции в Африке, у Массиева в Дакарском Университете, со своим люминисцентным микроскопом МЛ-2 и смотрели человеческие гепатомы ИФ-методом. Основной трудностью здесь было поглощение αF из крови мертвыми и гибнущими клетками. Гибнущие клетки в опухоли поглощали сывороточные белки и отличить поглощение из крови от внутриклеточного синтеза на срезах не представлялось возможным. Тогда мы ввели γ-глобулиновый контроль на серийных срезах, показывающий, какие клетки на срезе поглощают заведомо не синтезирующиеся в них белки, а какие – содержат αF строго специфически (см. «Методы» и Engelgardt et al.,1971). Эти опыты также дали вполне однозначные результаты – далеко не все клетки в гепатомах синтезируют αF, и уровень продукции опухолью αF коррелирует с числом αF-положительных клеток (Goussev et al. 1971). Эти данные вполне соответствовали гипотезе субпопуляций, но отнюдь не доказывали ее.
В этот период 1969–1970 гг. я готовил большой обзор по αF в Adv. Cancer Res., куда был приглашен его главным редактором Дж. Клейном (George Klein) осенью 1969 г. Туда вошли все работы по αF – его биологии и клинике, – опубликованные к тому времени (Abelev, 1971). Обзор вошел впоследствии в «Citation Classics» за период 1970–1985 гг. (Abelev, 1987). Подготовка этого обзора позволила нам держать перед собой всю картину исследований по αF и проанализировать все, что к тому времени было известно в этой области.
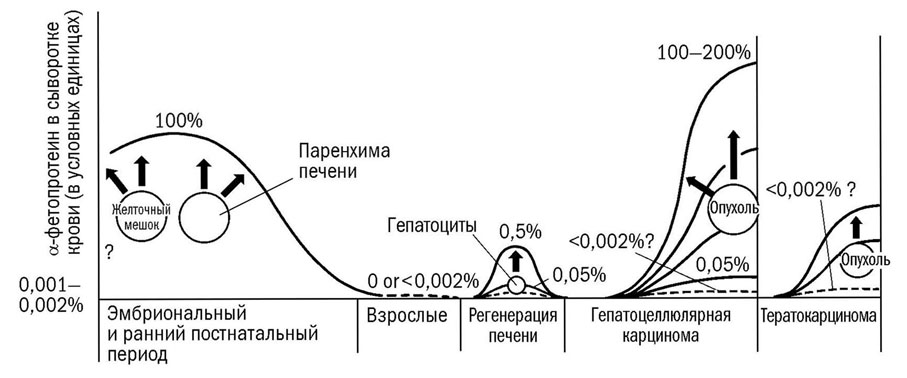
Схематическое изображение динамики АФП в онтогенезе и канцерогенезе. Сплошные линии – cывороточный уровень АФП, пунктир – ожидаемый уровень АФП, «?» – неизвестно (Абелев, 1971) |
Для дальнейшего анализа гипотезы мы воспользовались методом «обратного гемолиза в геле» (см. «Методы»). И первым подходом было изучение продукции сывороточного альбумина и АФП (6) в онтогенезе (Эрайзер и др., 1977 ).
Четкое перекрытие продукции обоих белков в популяции фетальных гепатоцитов человека говорило против клональной продукции сывороточных белков печенью (Эрайзер, 1987). Это перекрытие было подтверждено позже и иммуногистохимической локализацией альбумина и трансферрина (Энгельгардт, 1984; 1986). Таким образом, предположение о клональной структуре гепатоцитов, по крайней мере, в отношении белков сыворотки подтверждения не получило. Гепатоцит предстал как полифункциональная клетка с дифференциальной регуляцией секретируемых белков. Оставалась наиболее сложно анализируемая часть гипотезы – синтез АФП терминально дифференцированными гепатобластами в популяции опухолевых клеток. Для этого необходимо было исследовать клоногенную способность АФП-продуцирующих и непродуцирующих клеток в опухолях. После многолетних неудачных попыток нам удалось это сделать, используя метод обратного локального гемолиза в геле в сочетании с прослеживанием единичных клеток, охарактеризованных по синтезу АФП (см. «Методы», Эрайзер и Абелев, 1984; Эрайзер и др. 1987; Eraiser & Khamzina, 1988). Результаты были однозначными: АФП-положительные и АФП-отрицательные клетки крысиной гепатомы были в равной мере клоногенны – они давали колонии клеток, свидетельствующие о 5–7 генерациях. И хотя каждое из этих положений при дополнительных, ad hoc, допущениях могло быть совмещено с гипотезой –
смысла в этом не было, гипотеза становилась для фактов прокрустовым ложем. Вместе с тем гипотеза сослужила свою службу – она привела к обнаружению гетерогенности в нормальной и опухолевой локализации АФП, градиентного затухания синтеза АФП в постнатальной печени и дискретных «АФП+» клеток при регенерации печени, а также к установлению полифункциональности гепатоцитов и клоногенности «АФП+» и «АФП-» опухолевых клеток.
Новая версия гипотезы о причинах реэкспрессии АФП в гепатомах стала созревать еще в период работы над гипотезой предыдущей. В 1971 г. Китагава (Kitagawa) в Японии и Кроес (Kroes) в США показали, что в процессе химического гепатоканцерогенеза у крыс возникает высокая волна АФП, затем спадающая. У этих крыс впоследствие возникает гепатоцеллюлярный рак (ГЦР). Существовала определенная позитивная корреляция между появлением АФП и последующим развитием ГЦР. Эта волна четко коррелировала с появлением и пролиферацией овальных клеток – предшественников гепатоцитов и холангиоцитов, как бы стволовых клеток печени. Овальные клетки имели очень характерную морфологию – небольшие размеры, овальную форму, «изрезанное» ядро и неразвитый эндоплазматический ретикулум. Эти клетки дифференцировались в гепатоциты, проходя стадию юных гепатоцитов, и в холангиоциты. Банников и Чипышева из лаборатории Ю.М. Васильева обнаружили АФП в овальных клетках прямым иммунофлуоресцентным методом.
Появление овальных клеток происходило в тех случаях, когда зрелые гепатоциты были повреждены и не могли восстанавливать печень, а регенерация печени происходила за счет новообразования гепатоцитов. Было естественно предположить, что при канцерогенном воздействии, когда зрелые гепатоциты дегенерируют и не могут делиться, мобилизуется резерв восстановления печени – овальные клетки, которые и служат «популяцией высокого риска» для возникновения гепатом. Гепатомы, возникшие из овальных клеток, согласно гипотезе продуцируют АФП, как их нормальные предшественники. При этом они сохраняют способность овальных клеток к дифференцировке в гепатоцит, утрачивающий эту способность. Отсюда гетерогенность клеток в гепатоме, которая определяется различными стадиями их дифференцировки. Отсюда и редкие смешанные формы гепато- и холангиоцеллюлярного рака печени. Особенность синтеза АФП при регенерации печени, например, низкий синтез при гепатэктомии и значительно более высокий при действии CCl4 – легко объясняется пролиферацией овальных клеток при отравлении гепатоцитов CCl4 и регенерацией печени за счет деления только зрелых гепатоцитов при гепатэктомии.
Экспериментальные следствия, вытекающие из гипотезы – синтез АФП в овальных клетках при регенерации печени и в овально-клеточных элементах рака печени.
В этот же период (1968–1971 гг.) мы провели серию исследований по иммунодиагностике гепатом и тератобластом высокочувствительным методом иммунорадиоавтографии (см. «Иммунодиагностика») и исследовали причины продукции АФП тератобластомами (ТБ). В этом последнем случае мы исходили из данных Гитлина и сотр. (Gitlin et al. 1967, 1968) о том, что первым местом синтеза АФП и других сывороточных белков в онтогенезе является энтодерма желточного мешка (ЭЖМ), и Тейлума (Teilum, 1965; 1968) – о дифференцировке ТБ яичка и яичников в энтодерму желточного мешка. Вполне логично было ожидать, что АФП в ТБ синтезируется структурами, аналогичными ЭЖМ. Это было с полной убедительностью показано Н.В. Энгельгардт, В.С. Полтораниной и А.К. Язовой на мышиных тератобластомах (Engelgardt et al., 1973). Эти опухоли наряду с бесформенной массой эмбриональных клеток, способных к полипотентным дифференцировкам, образуют так называемые эмбриоидные тельца. Эти тельца аналогичны раннему эмбриону, содержащему эмбриональные недифференцированные полипотентные клетки, окруженные монослоем ЭЖМ. Энгельгардт, Полторанина и Язова абсолютно четко показали, что АФП обнаруживается иммуногистохимически только в клетках, соответствующих ЭЖМ, и что в эмбриоидных тельцах накапливается жидкость, сходная по составу с ранней сывороткой эмбриона, где преобладающим компонентом является АФП, хотя в окружающей асцитной жидкости был совершенно другой белковый состав с главным компонентом – сывороточным альбумином. Таким образом, мы впервые показали, что синтез АФП при ТБ обусловлен элементами ЭЖМ, сохраняющими свою нормальную функцию, а не самими эмбриональными клетками и не их дифференцировкой в нормальную печень.
Все данные по продукции АФП в норме и опухолях, по его диагностике и овально-клеточная гипотеза продукции АФП при ГЦР были представлены и проанализированы в больших обзорах (Abelev, 1974; Abelev et al., 1974). В это же время, независимо от овально-клеточной гипотезы, Гусев и Язова сделали интересную работу по преодолению толерантности к АФП (Goussev & Yazova, 1974). Они показали, что при иммунизации крыс мышиным АФП (но не крысиным), преодолевается толерантность к АФП крысы: в сыворотках иммунных животных обнаруживаются антитела, реагирующие с АФП мыши и крысы, а в перитонеальном эксудате методом подавления миграции макрофагов обнаружены Т-клетки, реагирующие как с иммуногеном, так и с собственным АФП крысы (Yazova et al., 1978). Одновременно, Nishi из Университета Хоккайдо показал, что кролики, иммунизированные человеческим АФП, отлично реагируют с кроличьим АФП.
Надо сказать, и не только сказать, а очень и очень подчеркнуть, что исследования в этот период (1971–1977 гг.) шли в обстановке травли и полной изоляции от международной науки.

Обложка номера Cancer Research, август 1974 |
Lev Alexandrovich Zilber (1894–1966), doctor of medical sciences and member of the U.S.S.R. Academy of Medical Sciences, was the founder of the Russian school of viral oncology. A graduate of Moscow State University in 1919, his earlier experimental work was on auto-serotherapy of typhus (1921), hereditary transformation of serotypes in Proteus vulgaris (1922–1923), and the replication of viruses in unnatural hosts, as vaccinia virus in yeast (1932–1934). He and his co-workers identified the tick-borne, summer-spring encephalitis of the Far East regions of the U.S.S.R. He began work on the virological aspects of cancer in 1944, heading the Department of Immunology and Virology of Tumors at the Gamaleya Institute of Epidemiology and
Microbiology, Moscow. [N. F. Gamaleya (1859–1949), after whom the Institute is named, was a bacteriologist who, following a visit with Pasteur in 1896, introduced rabies vaccination in Russia.] G. I. Abelev (b. 1928) doctor of biological sciences and professor of biochemistry, graduated from Moscow State University in 1950. He was an assistant of Dr. Zilber, whom he succeeded as departmental chairman at the Gamaleya Institute in 1966. Abelev and his colleagues devoted their attention to tumor-specific antigens and demonstrated striking immunological individuality in mouse hepatomas (cf, Progr. Exptl. Tumor Res., 7: 104, 1965). We are indebted to Dr. Lev L. Kisselev for the portraits of Zilber (left) and Abelev (right). |
Меня не только не выпускали из страны, но просматривали всю переписку, контролировали все контакты с иностранцами, не выпускали из Института даже для чтения лекций в Университете или для участия в работе оргкомитета Онкологического съезда (1972). Причина этого была та же, о которой я написал в разделе «Иммунодиагностика», а события частично описаны в очерке о В.А. Энгельгардте (Абелев 1993) и в главе 2 второй части этой книги «Драматические страницы…». Эти события привели нас после очень тяжелой борьбы в 1977 г. в Онкоцентр.
Что касается феномена АФП, то главным путем анализа следствий овально-клеточной гипотезы стал иммуногистохимический анализ регенерации печени, вызванной CCl4. Здесь можно было ожидать, что АФП будет обнаружен в отдельных клетках, отличных от гепатоцитов и относящихся к овальным или их ближайшим потомкам.
Данные, полученные Н.В. Энгельгардт, В.С. Полтораниной и сотр., а затем неоднократно и в разных условиях воспроизведенные, действительно показали, что АФП локализован (продуцируется?) только в отдельных клетках, граничащих с некрозом, образующимся под действием CCl4 вокруг центральных вен. У мышей BALBc/J и SWR, отличающихся повышенным фоновым уровнем АФП в крови и «высоким» ответом при регенерации печени, эти АФП-положительные клетки образовывали сплошной «монослой» вокруг некроза (Энгельгардт и др., 1976; Engelhardt et al., 1976; 1979). Можно ли было считать эти клетки «овальными»? Можно ли было считать их синтезирующими АФП или накапливающими этот белок? Можно ли было считать их вполне жизнеспособными? Ясно, что все наши экспериментальные исследования были сфокусированы на эти клетки. Н.В. Энгельгардт привлекла к нашим исследованиям В.Н. Баранова, первоклассного и единственного иммуноэлектронного микроскописта, работавшего тогда в Институте морфологии человека. М.Н. Лазарева, аспирантка, стала изучать действие разных гепатотоксинов на индукцию «перинекротического» эффекта и воздействие повторных волн регенерации на АФП-продуцирующие клетки, а В.С. Полторанина – изучала АФП-положительные клетки при многократном воздействии CCl4.
Эти исследования дали вполне определенные, хотя и не вполне ожидаемые результаты:
В.Н. Баранов абсолютно убедительно показал, что перинекротические клетки являются типичными зрелыми гепатоцитами, синтезирующими АФП (синтез подтверждался локализацией АФП на мембранах эндоплазматического ретикулума). Кроме того, он впервые выявил у мышей типичные овальные клетки, синтезирующие АФП, наряду с типичными зрелыми гепатоцитами (Баранов и др., 1981; 1982; Baranov & Engelgardt, 1987)
М.Н. Лазарева показала, что перинекротическая локализация АФП-положительных клеток не зависит от положения некроза в печеночной дольке: перицентральный или перипортальный некроз в равной мере был окружен валом АФП-положительных гепатоцитов. Овальные же клетки, как было известно из литературы, в основном локализуются в портальном районе дольки. Складывалось впечатление, что любые гепатоциты, независимо от положения в дольке, способны к реэкспрессии АФП (Лазарева, 1977).
Многократная регенерация печени, вызванная CCl4. ведущая к повторным некрозам и повторным волнам пролиферации, приводила к образованию гигантских полиплоидных гепатоцитов. И эти гепатоциты также экспрессировали АФП, если находились в перинекротической зоне (Лазарева, 1977; 1982; Engelhardt et al., 1979). Эти гигантские клетки и по морфологии и по происхождению, несомненно, относились к гепатоцитам.
Многократное воздействие CCl4 по схеме, ведущей к гепатоканцерогенезу, наоборот, вызывало пролиферацию мелких АФП-положительных клеток, природа которых – диплоидные юные гепатоциты или овальные клетки – установлена не была (Полторанина неопубл. – цитирую по Abelev, 1979).
Таким образом, вся эта серия экспериментов однозначно показала, что АФП при регенерации печени у мышей, получавших гепатотоксины, реэкспрессируется в зрелых гепатоцитах и, отчасти, в овальных клетках (последнее – у мышей с генетически «высоким» фоном АФП). Возникали ли эти АФП-положительные гепатоциты из предшественников (например, из овальных клеток, что очень мало вероятно), или синтез АФП возобновлялся в предсуществующих гепатоцитах?
Первые доказательства возобновления синтеза в предсуществующих гепатоцитах были получены Н.В. Энгельгардт и сотр. в совместной работе с лабораторией В.Я. Бродского (Ин-т биол. развития АН СССР). В опытах где одновременно выявлялся АФП с помощью ИФ и синтез ДНК по включению Н³-тимидина на срезах они показали, что синтез АФП в гепатоцитах начинается до вступления их в S-фазу (Engelhardt et al. 1976). Здесь были обнаружены клетки, в которых начался синтез АФП до включения Н³-тимидина. Далее М.Н. Лазарева, блокируя синтез ДНК гидроксимочевиной in vivo при непрерывном введении Н³-тимидина, показала, что синтез АФП после отравления CCl4 разворачивается по стандартной кривой в предсуществующей (не прошедшей деления) популяции гепатоцитов. Таким образом, вновь и другим методом было показано, что синтез АФП возобновляется в предсуществующих гепатоцитах и что критическим фактором является их перинекротическое расположение (Lazareva, 1981).
Очень четким и независимым доказательством возобновления синтеза АФП в предсуществующих зрелых гепатоцитах стали опыты В.С. Полтораниной по индукции синтеза АФП в печени при минимальных ее повреждениях. Небольшой (1-2 мм) надрез края печени приводил к небольшому некрозу без сигнала к регенерации. При этом вокруг некроза, захватывая и центральную вену, возникал очень четкий слой АФП-положительных клеток, микроскопически типичных гепатоцитов. Насыщающая Н³-тимидиновая метка, дававшаяся непрерывно в течение эксперимента, совсем не включалась в гепатоциты, в том числе, в гепатоциты перинекротического слоя. Следовательно, и в этих экспериментах, реэкспрессия АФП имела место в предсуществующих зрелых гепатоцитах (Полторанина, 1981). Стало ясно, что репрессия АФП в гепатоцитах обратима, и что клеточные взаимоотношения, по-видимому, являются критическими в контроле синтеза АФП.
В это время на меня сильное впечатление произвела статья Г.Н. Крыжановского о значении нервно-мышечных контактов в дифференцировке мышцы. Оказалось, что денервация мышцы ведет к ее дедифференцировке, а восстановление контакта ведет к реверсии дифференцировки. Те же отношения могли существовать и в печени. Вхождение гепатоцита в структуру печеночной балки вело к индукции и поддержанию его дифференцировки. Выход из балки, что наблюдалось, в перинекротической зоне, – вел к утрате гепатоцитом дифференцировки и к реэкспрессии АФП. В перинатальной печени балка «росла» от перипортальной области к центральной вене и так же устанавливался градиент супрессии АФП. На этой основе была сформулирована новая гипотеза – «структурной репрессии», которую я намеревался изложить на Гордоновской конференции по дифференцировке (1976 г.), куда был приглашен, но поездка туда разрешена не была (Abelev, 1978). Гипотеза «структурной репрессии» легла в основу наших последующих исследований. Анализ предыдущих данных и предпосылки гипотезы были обсуждены в детальных критических обзорах (Абелев 1979; Abelev, 1980).
Маркером «гепатоцита в структуре» служил для нас антиген желчных капилляров (АГ-1) (7), с которым мы много работали раньше (см. «Органоспецифические антигены»). АГ-1 был очень строго локализован в гепатоците в районе желчного капилляра, в апикальной части печеночной клетки. Он появлялся в гепатоците только тогда, когда гепатоцит встраивался в печеночную балку и образовывал желчный капилляр и, потому, служил маркером «структурированного» гепатоцита, находящегося в балке. В постнатальной печени АГ-1 обозначал построившуюся балку, а в отравленной CCl4 – его отсутствие – районы разрушенной балки. Серийные срезы постнатальной или регенерирующей печени давали возможность обнаружить АГ-1 и АФП в одних и тех же клетках.
А.С. Глейберман, наш гистолог, виртуозно красящий и тонко чувствующий клетки, профессионал клеточной биологии, проводил эту работу. По мнению Глейбермана перинекротическая позиция АФП-положительных клеток была связана с приобретаемой ими способностью активно внедряться, как бы двигаться, в зону некроза (Глейберман и др. 1979; 1980). С этим предположением хорошо согласовались его опыты с С.М. Трояновским по перестройке актинового скелета в краевых перинекротических клетках (Глейберман и др., 1984).
Мне же казалось весьма вероятным, что макрофаги, накапливающиеся в зоне воспаления и секретирующие протеазы (в том числе коллагеназу), могут способствовать выходу гепатоцита из балки. Это можно было проверить блокируя протеазы, но до этих опытов мы, к сожалению, не дошли.
Тонкое исследование постнатальных гепатоцитов и гепатоцитов в перинекротическом слое отравленной CCl4 печени мышей показало, что в первом случае АФП затухает в клетках, входящих в печеночную балку, и этим определяется градиент АФП вокруг центральной вены. «Кольцо» АФП-положительных клеток вокруг центральной вены состояло из АГ-1-отрицательных клеток. Во втором случае, особенно демонстративном, клетки перинекротического слоя были концевыми клетками балки, «смотрящими» в некроз. Они постепенно утрачивали АГ-1, сначала на одном конце, потом на другом, теряли связь с балкой, актиновый скелет их перераспределялся по эмбриональному типу, и клетка, как бы, внедрялась в некроз. При этом в ней появлялся АФП (Глейберман и др. 1979; Gleiberman et al., 1983; Глейберман и др., 1984). Интересно, что у «низко-индуцибельных» мышей (С57BL) краевая перинекротическая зона оставалась жестко связанной с балками, не внедрялась в некроз и не реэкспрессировала АФП (Глейберман и Полторанина, 1984).
Как мы уже говорили выше, клетки перинекротического слоя были типичными зрелыми гепатоцитами (Baranov & Engelgardt, 1981). Так были получены первые подтверждения «структурной репрессии АФП» и роли клеточных взаимодействий в регуляции синтеза АФП. Вскоре стало все более и более ясным, что речь здесь идет об общем феномене – роли положения клетки в печеночной балке в формировании ее фенотипа, в том числе, эмбрионального (Глейберман, 1980; Gleiberman & Abelev, 1985). Спустя несколько лет группа нидерландских эмбриологов пришла к тем же выводам, показав, что постнатальное затухание АФП определяется «архитектурной супрессией», т.е. строительством печеночной балки (Moorman et al., 1990; Notenboom et al. 1996). Особенно красиво они показали это на трансплантатах АФП-положительных клеток в печень in vivo (Notenboom et al. 1996) (8).
Можно было ожидать, что тот же принцип «супрессии» сохраняется в высокодифференцированных гепатомах. И, действительно, краевой надрез крупного гепатомного узла в печени приводил к некрозу и перинекротическому появлению АФП (Шипова и Глейберман, 1984; Shipova & Gleiberman, 1985).
Следующий принципиальный шаг был сделан А.С. Глейберманом, показавшим «вспышку» реэкспрессии АФП в изолированных зрелых гепатоцитах мышиной печени in vitro (Глейберман, 1982). Реэкспрессия АФП была близка к 100% по числу синтезирующих его клеток.
Далее, Глейберман, медленно вращая суспензию гепатоцитов в чашке Петри, получал «островок» клеток с плотным центром и разреженной периферией. В плотном центре АФП не реэкспрессировался, а вся периферия охватывала «отрицательный» центр АФП-положительным кольцом. Клетки вцентре объединялись щелевыми контактами в единую систему. АФП-положительные клетки по периферии не имели щелевых контактов (Глейберман и др., 1987; Gleiberman et al., 1989a).
Далее, изолированные клетки высаживались в чашки, дно которых было покрыто высушенным коллагеном I типа – они реэкспрессировали АФП. Клетки, высаженные между двумя слоями коллагена, собирались в балко-подобные структуры, с четко очерченными желчными капиллярами, маркированными АГ-1. Клетки внутри всей «балки» были связаны щелевыми контактами, АФП в них был полностью супрессирован.
Аналогичный эффект наблюдался при совместном культивировании гепатоцитов с непаренхимными клетками печени мыши (IAR) (9) – гепатоциты собирались в плотные островки, заключенные во внеклеточный матрикс, покрывающий островки сверху и подстилающий их снизу. Совместное культвирование гепатоцитов с IAR'ами было предложено Guillоuzo и Guillоuzo во Франции для сохранения нормальных функций гепатоцитами, используемыми для тестирования фармакологических препаратов. А.С. Глейберман посещал лабораторию Guillouzo, где и познакомился с этим методом.
Совместное культивирование гепатоцитов с IAR'ами было особенно эффективным. Вся эта серия экспериментов четко показала, что переключение с АФП-позитивного в АФП-негативное состояние гепатоцитов контролируется внеклеточным матриксом (ВКМ), создающим трехмерный каркас для клеток (Gleiberman et al., 1989 b; Кудрявцева, 1992; Kudrjavtseva & Gleiberman, 1994).
Мы предположили, что существует 2 стабильных состояния зрелого гепатоцита: поляризованное, характеризующееся кубоидальной формой, наличием апикального, латерального и базального доменов, желчных капилляров, экспрессией АГ-1 и супрессией АФП, и альтернативное состояние – деполяризованные клетки, распластанные в культуре, лишенные доменов, желчных капилляров и АГ-1 и экспрессирующие АФП. ВКМ контролирует переход из деполяризованного состояния в поляризованное, а разрушение ВКМ – из поляризованного в деполяризованное. Очевидно, что ВКМ в этих случаях контролирует не обособленно экспрессию АФП-гена, а все состояние гепатоцита, в котором АФП составляет лишь один признак (Abelev, 1993; см. Abelev & Eraiser, 1999).
К сожалению, А.С. Глейберман после долгой стажировки на рабочих местах в Штатах стал там работать, оставив исследования по АФП на пике интереса к ним, а в лаборатории продолжала это направление его аспирантка, Е.И. Кудрявцева, которая после защиты (очень успешной) и длительного лаг-периода, воспроизвела систему гепатоциты-IAR'ы и показала, что IAR'ы, трансформированные RAS'ом как и спонтанно-трансформированные, не подавляют синтез АФП в диспергированных гепатоцитах. Они строят дефектный ВКМ, который не создает трехмерного каркаса, покрывающего гепатоциты сверху. Гепатоциты не собираются в печеночно-подобные островки и продолжают синтезировать АФП (Kudrjavtseva et al., 2003). К сожалению, и Кудрявцева отправилась в Штаты, прекратив свою работу. В таких отъездах заключается драма нашей науки, а может быть, и самих отъезжающих (Абелев, 1999).
Нерешенным оставался вопрос о причине возобновления продукции АФП в гепатоцеллюлярном раке (ГЦР). Одна из гипотез, уже обсуждавшаяся, – овально-клеточная природа ГЦР. У этой гипотезы есть серьезные сторонники, например, Hixon и Sell в США. Возможно, что какая-то часть гепатом имеет овально-клеточную природу, но основательно доказанных случаев такой природы для ГЦР нет. Большинство гепатом имеет очевидное гепатоцеллюлярное происхождение по своей морфологии и гепатоцитарным маркерам, особенно у мышей (см. раздел «Органоспецифические антигены»). Более того, пристальное рассмотрение биологии гепатоцитов ясно выявляет в них сочетание признаков полустволовой и зрелой дифференцированной клетки (Абелев, 1983). Гепатоциты способны практически к неограниченной пролиферации и они чувствительны к регуляторным воздействиям, в том числе, к ростовым факторам, например, HGF (Hepatocyte Growth Factor). Они «чувствуют» регенераторный стимул и вступают в необходимое число делений при восстановлении печени. Другими словами, они обладают всеми признаками полустволовой клетки и одновременно выполняют функции зрелой, полностью дифференцированной клетки. Следовательно, они относятся именно к тому типу клеток, который, как правило, является предшественником опухоли и они сами, весьма вероятно, могут служить исходной клеткой для возникновения гепатомы (см. Абелев, 1982).
Наша позиция в сравнении опухолевой и нормальной ткани состояла в том, что «опухоль сохраняет направление и уровень дифференцировки нормальной клетки-предшественницы», но с допущением дальнейшего снижения этого уровня в процессе прогрессии (Абелев, 2000; Abelev & Lazarevich, 2006). Интересным подтверждением дифференцировочного статуса гепатом являются опыты Н.И. Куприной и др., (1993) по индукции дифференцировки в гепатобластомной линии человека Hep G2. Эта опухоль, активно продуцирующая АФП, при росте в присутствии DMSO выстраивала органотипичные островки (10) с полной супрессией АФП (Куприна и др., 1993).
Как объяснить с позиций нормальной регуляции АФП его реэкспрессию в опухолях? Другими словами, как связать возникновение ГЦР с блокированием взаимодействия гепатоцита с ВКМ? Хорошо известно, что процесс канцерогенеза применительно к сoлидным опухолям, т.е. карциномам и саркомам, включает два элемента – инициацию и промоцию. Инициация – это собственно опухолевая трансформация (мутация онкогена или гена-супрессора), в то время как промоция – это снятие тормозящего (нормализующего) влияния окружающей нормальной ткани. Это влияние осуществляется через взаимодействие клетки с ВКМ, поддерживающим ее дифференцированное состояние через межклеточные контакты, и TGF-β, тормозящий пролиферацию эпителиоцитов. Следовательно, нарушение взаимодействия с ВКМ необходимо для роста очагов трансформации и их превращения в карциному – автономно растущую и инвазивную. Это нарушение может осуществляться через мутации в интегриновой системе, осуществляющей связь клетки с ВКМ, либо через нарушение ВКМ-продуцирующих клеток стромы. Нарушение взаимодействия с ВКМ ведет к реэкспрессии в трансформированном гепатоците АФП, что является по нашему мнению, эпифеноменом, связанным с прогрессией опухоли, а не с собственно злокачественной ее трансформацией (Abelev, 1999; Abelev & Eraiser, 1999; Abelev & Lazarevich, 2006).
Молекулярные аспекты регуляции АФП
Наш анализ регуляции АФП все более уходил в изучение клеточных механизмов этого процесса. Тем временем, начиная с конца 70-х годов, стало развиваться изучение молекулярной генетики АФП – структуры его гена и молекулярных механизмов регуляции (Belanger, Sala-Trepat, Tilghman, Tamaoki и др.). К началу 80-х годов стала известна структура гена АФП человека и крысы, а вскоре и структура регуляторного района гена АФП. Этот район включал три тканеспецифических энхансера и промотор, а также сайленсер – участок, примыкающий к промотору, необходимый для подавления активности гена АФП (Vaсher & Tilghman, 1990) (см. Abelev and Lazarevich, 1996; Лазаpевич, 2000).
В последнее десятилетие было идентифицировано несколько «положительных» транс-факторов, взаимодействующих с энхансерами и промотором гена АФП и определяющих его активацию, в том числе «позиционную», т.е. проявляющуюся в разных участках печеночной дольки. Транс-фактор, взаимодействующий с сайленсером идентифицирован не был. Молекулярные механизмы постнатального подавления экспрессии гена АФП в постнатальной печени, равно как и причины реэкспрессии гена в опухолях, установлены не были. Поэтому первоочередным и определяющим казался вопрос, как ВКМ передает сигнал к подавлению синтеза АФП (Abelev, 1987, 1988). Для ответа на этот вопрос необходимы были молекулярные исследования, и мы начали думать об их организации. Мы установили рабочий контакт с группой А.В. Гудкова, передав ему 2-х сотрудников, молекулярных биологов – К.Н. Кашкина и Л. Третьякова, а также аспирантку Н.Л. Лазаревич, взятую специально для молекулярных работ по АФП. Впоследствии А.С. Глейберман, «главный человек», установивший роль ВКМ в регуляции АФП, был послан в США, в Калифорнийский Университет к Х.Лефферту для обучения молекулярным подходам к регуляции АФП.
Гудков уехал в США, Глейберман, продлив пару раз пребывание у Лефферта, остался с семьей в Сан-Диего, а с нами продолжала работать аспирантка Гудкова Н.Л. Лазаревич, развивая все бóльшую активность. При ней сложилась отличная молодежная молекулярная группа (Е.В. Варга, О.А. Черемнова, Д.А. Овчинников и И.Ф. Кустова). Они сконцентрировались на сравнении «АФП+» и «АФП-» клонов, выделенных Т.Л. Эрайзер из крысиной гепатомы МсARH 7777. Клоны отличались стопроцентно по продукции АФП и их сравнивали по нескольким параметрам.
Во-первых, был применен гибридомный анализ с целью получить МкАТ, специфически реагирующие только с «АФП-» клоном, и это нам удалось. Одно из антител (МкАТ А2/3) к «АФП-» клону 7Е10 выявляло антиген (Аг А2/3) только в АФП-отрицательных клонах и не реагировало с «АФП+» клетками независимо от того, были ли это «АФП-» клоны гепатомы McARH 7777 или других «АФП-» линий. Напротив, антиген А2/3 отсутствовал в «АФП+» штаммах и «АФП+» гепатоме Зайдела. Антиген А2/3 отсутствовал также в нормальной печени, но легко и эффективно индуцировался в ней солями Pb++ и Cd++. Cd++ индуцировал также Аг А2/3 в «АФП+» клоне исходной гепатомы (Eraiser et al., 1998). Аг А2/3 по мол.весу (~ 45 кД) и по индукции температурой отличался от белков теплового шока. Таким образом, гибридомный анализ выявил в гепатоме 7777 белок клеточного стресса с альтернативной экспрессией по отношению к АФП. В последующий год наши «молекулярщики» старались получить экспрессирующую библиотеку mРНК из «АФП-» клона, клонировать ее в E.coli и экспрессировать в бактериальной системе. Эта попытка не удалась, и мы переключились на очистку Аг А2/3 иммунохимическим методом, и эта работа в настоящее время в ходу. Она оказалась, однако, столь трудна, что до сих пор (2005–2006 гг.) мы приближаемся к очистке АГ А2/3, не получив его еще в достаточно чистом виде, пригодном для секвенирования.
Второй подход – преимущественно молекулярный, при котором анализировались в альтернативных клонах специфичные для печени транс-факторы HNF-1, HNF-3 и HNF-4. Было обнаружено, что HNF-4 резко (на два порядка) снижен в «АФП-» (по mРНК HNF-4), что ведет и к снижению HNF-1. Трансфекция «АФП-» клона плазмидой, содержащей HNF-4, вела к его реверсии по экспрессии HNF-4 и HNF-1, но не к продукции АФП (Лазаревич, 1997; 2000; Lazarevich et al., 1998; 1999).
Третий подход – соматическая гибридизация «АФП+» и «АФП-» клонов, которая четко показала доминирование супрессии АФП-гена. (Кустова, 2000; 2001). Мы надеемся в этой системе идентифицировать «негативный» транс-фактор, участвующий в регуляции АФП-гена.
Так или иначе, эстафета в исследовании регуляции АФП переходит в нашей лаборатории в руки молекулярной группы. Направление работ этой группы все дальше уходит в изучение связи специфичных для печени HNF с процессом опухолевой прогрессии (Лазаревич, 2004; Lazarevich et al., 2004; Abelev & Lazarevich, 2006).
Сомкнется ли это направление с выяснением молекулярных механизмов регуляции АФП?
Эпитопная карта и эпитопные варианты АФП
Как я уже писал раньше (см. «Методы») в 1980 г. в лаборатории была получена коллекция из 15 МкАТ к АФП человека и 3 МкАТ к мышиному АФП, которые были охарактеризованы по перекрестным реакциям друг с другом (Язова и др., 1982; Goussev et al., 1990; Yazova et al. 1990). МкАТ к человеческому АФП предназначались для РИА наборов, совместно с ташкентским «Радиопрепаратом», но распад Союза и, соответственно, спад спроса на новые диагностические препараты привел к тому, что широкое производство моноклонального набора не началось. Эти МкАТ были охарактеризованы А.В. Андреевым по сродству к АФП и некоторые из них были использованы для создания собственного лабораторного РИА – набора (Андреев и др., 1993; Андреев и Григорьева, 1996; 1998; 2001).
Но главная работа с МкАТ развернулась по эпитопному картированию АФП, в котором широко использовалась иммуноаффинная электрохроматография (ИАЭ) (см. «Методы»). Метод позволял автоматически и быстро (3-4 часа), качественно (т.е. по «+» или «-») и сразу на большом числе МкАТ определять перекрестную реактивность МкАТ к АФП или любому другому антигену. Мы применили ИАЭ сначала к небольшому числу МкАТ и легко установили отдельные и перекрестно-реагирующие эпитопы АФП (Christiansen et al.1994; Abelev et al., 1994). Затем, в 1996 г. была организована Международная рабочая группа по сравнению 30 МкАТ, собранных из различных лабораторий и компаний мира (Alpert & Abelev, 1998). Эти антитела были охарактеризованы по перекрестной реактивности с АФП в ИАЭ. Были определены 5 дискретных эпитопных кластеров (А, В, С, D, E) и выявлены родственные отношения между некоторыми из них (Yakimenko et al. 1998). Наши данные почти полностью совпали с результатами РИА (Nustad et al, 1998) (11), а расхождения касались МкАТ, которые не могли анализироваться в РИА из-за низкого сродства, но отлично определялись в ИАЭ (Эпитоп D и МкАТ С9). В последующие 2 года коллекция была дополнена еще 21 МкАТ и более полная и детальная карта была составлена Якименко (Якименко и др., 2001). Встал вопрос о проецировании эпитопной карты на первичную структуру человеческого АФП. Эта работа была проведена группой Nishi et al. в университете Хоккайдо (2001) (12) В этой работе были локализованы основные эпитопы полипептидной цепи АФП.
Полностью неожиданным оказалось выявление с помощью ИАЭ эпитопных вариантов человеческого АФП. Вначале мы разделили с помощью ИАЭ «D+» и «D-» варианты человеческого АФП, а затем и (А-В)+ и (А-В)- варианты. Мы предполагали, что здесь могли выявиться фрагменты АФП, отличающиеся по локализации эпитопов (Christiansen et al. 1994; Abelev et al. 1994). Затем мы предприняли систематические исследования эпитопных вариантов АФП, сочетая разделение вариантов АФП на «зебре» с их элюцией с МкАТ-слотов кипячением в 2% SDS, разделением в SDS-PAG и иммуноблоттингом. Таким способом мы показали, что обнаруженные варианты (А-В)+ и (А-В)-; Е+ / Е-; В+ / В-; А+ / А- и часть D+ / D- имеют идентичный мол. вес и являются конформационными вариантами АФП (Abelev et al., 2003, Karamova et al., 2001, 2003).
Применив более легкие условия элюции подфракций АФП с микросорбентов, мы получили лучшую сохранность эпитопов и смогли показать, что во всех эпитоп-отрицательных вариантах АФП можно было в иммуноблоттинге выявить «скрытые» эпитопы, недоступные антителам в нативной молекуле.
Таким образом, мы показали, что молекула АФП очень чувствительна к конформационным воздействиям и вызванные ими изменения четко выявляются по сдвигу равновесия эпитоп)+ /эпитоп)- вариантов.
Такова новая область исследования АФП, в которую мы вошли благодаря разработанному методу ИАЭ и построению с его помощью эпитопной карты АФП.
* * *
Таким образом, изучение АФП было начато нами, когда АФП еще был «специфическим антигеном гепатомы». Мы прошли через теоретические и практические этапы изучения проблемы и остановились в области «молекулярно-клеточных» механизмов его регуляции. Практические диагностические аспекты проблемы полностью или почти полностью были решены, а теоретические активно изучаются в этой системе, которая стала одной из наиболее четких и выразительных в проблеме регуляции генов в онтогенезе и канцерогенезе. Мы же все эти годы как раз и стремились вывести проблему регуляции АФП в сферу внимания клеточных и молекулярных биологов. Своим главным вкладом в проблему регуляции АФП мы считаем установление роли клеточных взаимодействий и роли ВКМ в регуляции гена АФП.
Основные задачи сегодня – понять как взаимодействие с ВКМ определяет тканеспецифический набор HNFs в гепатоцитах (и, возможно, в других эпителиальных органах) при индукции по поддержанию их дифференцировки.
Примечания
(1) На этой возможности настаивал А.Я. Фриденштейн – наш друг и постоянный, полный сарказма, критик. Назад
(2) С.Д. Перова пришла к нам к самому началу этой работы, поступив на «декретное» место Н.И. Храмковой (Куприной). Назад
(3) Эта работа вошла в коллекцию М. Shimkin (ed.) Some classics of experimental oncology: 50 selections, 1785–1965. NIH publication, 1980, pp. 601–607. Abelev, G. I., Perova, S. D., Khramkova, N. I., Postnikova, Z. A. and Irlin, I. S., Production of embryonal α-globulin by transplantable mouse hepatomas. Transplantation 1:174-180, 1963 Эмбриональный сывороточный альфа-глобулин и его синтез перевиваемыми гепатомами мышей. Г. И. Абелев, С. Д. Перова, И. И. Храмкова, 3. А. Постникова и И. С. Ирлин Назад
(4) Uriel J. In: IARC scientific publications N 1, Liver Cancer, 1971, pp. 58–68 Назад
(5) B. Spear: Semin. Cancer Biol. 1999, 9(2), pp. 109–116. Назад
(6) С 1970 г. αF стал называться альфа- фетопротеин (АФП). Назад
(7) В последствии идентифицированный с Bgp-1 (см. «Методы») Назад
(8) Notenboom et al. Development 122, 321–332, 1996 Назад
(9) IAR – клеточные линии из непаренхимных клеток печени, неустановленной природы, полученные в лионском Int. Agency of Research on Cancer (IAR) Назад
(10) Цитокератин менял эмбриональную (фибриллярную) локализацию на «взрослую» (подмембранную.) Назад
(11) Nustad et al. Tum. Biol. 19(2), 293–300, 1998. Назад
(12) Kang, G., Matsuura, E., Sakamoto, T., Sakai, M., and Nishi, B. (2001) Tumor Biol., 22(4), 254–261. Назад