ContentI. IntroductionII. Normal epitheliocyte A. Differentiation markers III. Microenvironmental control of transformed cells A. Control of transformed cells by normal surrounding IV. Concluding remarks A. Different ways of progression in hemoblastoses and carcinomas References |
I. Introduction
Differentiation state of malignant tumors is an important characteristic, which helps to establish their histological origin and to understand the degree of deviation from normal biology, as well as the stage in tumor progression and peculiarities in clinical behaviour. It offers the basis for search of tumor markers and investigation of their nature. Differentiation antigens are absolutely necessary for precise classification of hemoblastoses and, hence, for their diagnosis and prognosis. At present time the immunophenotyping is a routine procedure in hematological oncology (reviewed by Van Dongen et al., 2002). Much less is known about epithelial tumors and this review discusses that problem.
Malignant growth can be considered as a result of two successive events – cell transformation and tumor progression. Here we define transformation as autonomous proliferation of an immortal cell clone, while the progression as the process, leading the immortal transformed clone to invasion and metastasis.
Progression proceeds through continuing selection of the most autonomous cell variants from a genetically unstable population of transformed cells (Nowell, 2002). Progression is a stepwise endless process started in vivo in the transformed clone. Acquisition of malignancy is the result of tumor progression rather than of transformation.
Transformation itself is not necessarily associated with the loss of differentiation. Certain well differentiated hemoblastoses, such as plasmocytomas continuing immunoglobulins (Ig) production throughout their growth in primary host or during serial transplantations, might serve as an illustration of compatibility of transformation and malignancy with persistence of differentiation (Stevens and Craig, 1999). Another example is chronic myeloid leukemia associated with overproduction of mature granulocytes lacking the ability to enter apoptosis (Deininger et al., 2000). Maintenance of differentiation state in the hemoblastosis progenitor cell might be regarded as a rule for hemopoietic neoplasms (reviewed by Abelev, 2000).
In epithelial tumors, the progenitors (transformed cells) evolve during tumor progression and become more and more autonomous. In this process, the tumor cells change their morphology and behavior. They lose cuboidal shape and polarity and become more independent from neighbouring tissues. Finally, they acquire the capacity to invade the underlying tissue and to form distant metastases.
Tumor progression is usually associated with partial or complete loss of morphological and biochemical features of the original tissue, i.e. with dedifferentiation.
We assume that the tumor microenvironment plays an especially important role in maintenance of the differentiation state, and independence from the microenvironment is a crucial factor in dedifferentiation of tumor cells. We would try to substantiate this opinion in the present review.
It looks highly plausible that epithelial cell interactions with microenvironment are responsible for the differentiation state maintenance as well as for the control of behavior of transformed cells. The capacity to invasive growth and metastasis is acquired by transformed cells in the process of tumor progression, which leads simultaneously to gradual or stepwise liberation from microenvironment control and, hence, to partial or complete loss of their differentiation state.
II. Normal epitheliocyte
A. Differentiation markers
Epitheliocyte is a very peculiar cell with remarkable morphophysiological characteristics. It is cuboidal, or columnar in shape, polygonal and polar with distinct domains: apical and basolateral (Fig. 1). Polarized cells form acini with lumens inside the acini. The apical domain faces the lumen and is covered by glycoproteins, which belong to Ig superfamily: Bgp1 (biliary glycoprotein 1) in the apex of hepatocyte (Kuprina et al., 1990; Daniels et al., 1996) or CEA (carcinoembryonic antigen) along the apical part of intestinal cells (Hammerström et al., 1999). These glycoproteins are very clearly recognized by specific antibodies and are distinct markers of epitheliocyte (especially hepatocyte) polarity.
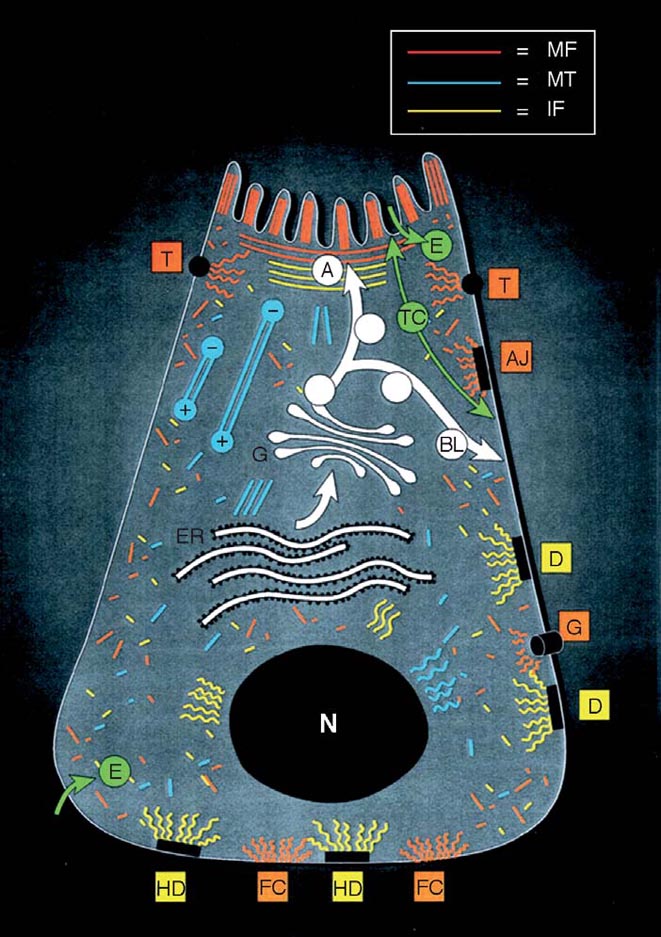 |
Figure 1. Schematic diagram of epithelial cell polarity, cell-cell, and cell-substratum junctions. Polarized epithelia, as found in the intestine, contain an apical domain with specialized features such as microvilli and a basolateral domain that are separated by tight junctions. Plasma membrane proteins reach their ultimate target domain by a direct or an indirect (transcytotic) pathway involving microfilaments (MF) and microtubules (MT). The apical and basolateral domains have distinct organization of underlying cytoskeleton. For example, the MT organizing center underneath the apical membrane generates a uniform polarity of MT with the apical minus and basal plus ends, allowing vesicle transport in two directions. Tight, gap, and adherens junctions and focal contacts link with actin filaments (attachment of MF with gap junctions is not well defined, which is indicated by the close proximity but not attachment to MF), whereas desmosomes and hemidesmosomes connect with intermediate filaments (IF). A, apical; BL, basolateral; T, tight junction; AJ, adherens junction; D, desmosome; G, gap junction; HD, hemidesmosome; FC, focal contacts; E, endocytosis; TC, transcytosis; N, nucleus; ER, endoplasmic reticulum; GA, Goldji apparatus (From Ku et al., 1999). |
The basal part of epitheliocyte is rich in integrins recognizing extracellular matrix (ECM), particularly proteins of basal membrane and fibronectin. The basal membrane is a common part of all epithelial tissues. It serves as a mechanical support of any epithelia and delimits the territory of epithelial tissue. The basal membrane supports the epithelial layer and determines the polarity of epitheliocytes (Brill et al., 2001). The basal membrane of different epithelial organs has different chemical composition: e.g. type I collagen in the tendons and type IV collagen in the liver, gastro-intestinal tract and kidney.
The lateral surfaces of epitheliocyte are rich in characteristic adhesion protein, E-cadherin, an essential component of adherence junctions and desmosomes, responsible for specific recognition of homologous neighbouring cells and for creating continuous intercellular network of tissue specific intermediate filaments, responsible for epithelial layer elasticity.
It is very important that cytoplasmic domains of E-cadherin are associated with α, β and γ-catenin molecules. When E-cadherin is downregulated or disrupted, the catenin complex is dissociated, β-catenin migrates into the nucleus and functions as a transcription factor (Cavallaro and Christofori, 2004).
E-cadherin has a very peculiar localization between the lateral surfaces of epitheliocytes in the epithelial layer and can be detected by the corresponding antibodies (Ku et al., 1999).
Specific proteins of gap junctions connexins (Cx) are also localized on the lateral surfaces on epithelial cells (Fig. 1).
The network of intermediate filaments is tissue specific and serves as a specific marker for epithelial classification and detection of remote metastases. Intermediate filaments localize in the cytoplasm and are connected with hemidesmosomes, located on basal membrane (Fig. 1).
Ig-like cell adhesion molecules are additional markers, which also could be visualized immunohistochemically.
Integrins are molecules of great importance, they recognize specifically ECM components, such as collagen of different types, fibronectin, and proteoglycans (Giancotty and Ruoslahti, 1998; Ruoslahti, 1999).
Additional markers of epitheliocytes are proteins synthesized by them, such as serum albumin, alpha-fetoprotein (AFP), transferrin, α1-antitrypsin or cytochrome P450 in hepatocytes, specific proteases and amylases in the pancreas, or casein in mammary gland epithelium (Ben-Zeev et al., 1988; Di Persio et al., 1991; Guillouzo et al., 1993; Schmeichel and Bissell, 2003).
Epitheliocytes (especially hepatocytes) possess a remarkable property to quickly and reversibly dedifferentiate after isolation and explantation on plastic, but maintain differentiated state in proper ECM. This property allows studying the dynamics of epitheliocyte markers expression during de- or re-differentiation of hepatocytes (Section III. B).
Studies of the expression patterns of epitheliocytes markers in transformation and progression can disclose elementary events associated with the evolution from transformation to malignancy.
B. Tissue-specific transcription factors
Transcription factors essential for expression of the majority of functional epithelial proteins are invaluable markers of tissue differentiation and its changes in the process of progression.
Tissue-specific gene regulation is best studied in the liver. The extensive studies of the last 15 years led to detailed characterization of liver-specific transcriptional network. Here we review the main properties of this regulatory cascade in hepatocytes and then discuss its relation to other epithelial organs.
1. Hepatocyte nuclear factors (HNF) in liver development and differentiation
The fine regulation of liver-specific gene expression and the maintenance of hepatic differentiation are mainly implemented by combinatorial action of hepatocyte nuclear factors (HNF) (Tronche and Yaniv, 1992; Locker, 2001). This class of proteins includes five families of transcriptional regulators (Table 1) with binding sites located in the majority of regulatory modules of liver-specific genes and required for their proper expression (for detailed reviews see Cereghini, 1996; Lazarevich, 2000; Schrem et al., 2002). The tissue-specificity of the expression of each hepatic gene is achieved by simultaneous participation of several HNFs in the regulation of this process.
| Gene (synonym) | Structure of DNA-binding domain | Embryonic expression (days of gestation) | Representative target genes | Pattern of expression |
| HNF1α (HNF1 LFB1, TCF1) | Variant homeodomain | 10.5 | AFP, albumin, α1-antitrypsin, tyrosine aminotransferase, transthyretin, aldolase B, apolipoproteins L2, T | Liver, intestine, kidney, pancreas |
| HNF1β (vHNF1, LFB3, TCF2) | 4.5 | Liver (low), kidney, intestine, pancreas, esophagus, thyroid | ||
| HNF3α (FoxA1, TCF3A) | Winged helix | 7.5 | Albumin, AFP, transthyretin, α1-antitrypsin, transferrin, apolipoprotein B, tyrosine aminotransferase, HNF1α, HNF3α, HNF3β | Liver, intestine, stomach, lung, pancreas, prostate |
| HNF3β (FoxA2, TCF3B) | 6.5 | Liver, intestine, stomach, lung, pancreas | ||
| HNF3γ (FoxA3, TCF3G) | 8.5 | Liver, intestine, stomach, testes | ||
| HNF4α (NR2A1) | Zink finger | 4.5 | HNF1α, transferrin, α1-antitrypsin, transthyretin, albumin, apolipoproteins A1, A2, B, C2, C3, aldolase B | Liver, intestine, pancreas, kidney |
| HNF6 (Onecut1) | Onecut | 9 | HNF1β, HNF3β, HNF4α, transthyretin,
α1-antitrypsin, glucokinase | Liver, pancreas, spleen, brain |
| C/EBPα | Basic region leucine zipper | 13 | AFP, albumin, apolipoproteins L1, L2, T, transthyretin, transferrin, tyrosine aminotransferase, type 1 collagen, metalloproteinases | Liver, intestine, lung, adipose, ovary, mammary gland, skin, skeletal muscle, placenta |
| C/EBPβ (LAP, NF-IL6, TCF5) | 12 | Ubiquitous, predominates in liver and lung |
HNF1 family includes factors HNF1α (Cereghini et al., 1988) and HNF1β (De Simone et al., 1991; Rey-Campos et al., 1991) binding the same DNA consensus as homo- or heterodimers with different trans-activation properties (reviewed in Tronche and Yaniv, 1992). HNF1 binding sites are abundant in the promoters of liver-specific genes (Tronche et al., 1997). While interacting with multiple coactivators that possess histone acetyltransferase activity, HNF1 proteins are able to alter the local chromatin structure and activate transcription (Soutoglou et al., 2000).
HNF1β is first detected in early embryonic development and is essential for visceral endoderm formation in mouse embryo as indicated by early embryonic lethality of HNF1β knockout mice (Coffinier et al., 1999; Barbacci et al., 1999). Liver-specific inactivation of the HNF1β induces defects in intrahepatic bile ducts and gallbladder formation (Coffinier et al., 2002). HNF1β expression is also detected at the onset of pancreatic exocrine ducts and kidney tubules formation (Coffinier et al., 1999).
The expression of HNF1α starts later, during liver specification, and its level is significantly lower than that of HNF1β (Ott et al., 1991). In the adult liver, this balance is changed and HNF1α predominates. Targeted disruption of HNF1α gene in mice does not generate considerable abnormalities in liver development, but causes progressive wasting syndrome, defects in insulin secretion, renal dysfunction, increase of liver mass and downregulation of some liver-specific genes during the first weeks after birth (Pontoglio et al., 1996; Pontoglio et al., 1998). It is likely that HNF1β plays a key role in organogenesis, while HNF1α is mainly involved in the maintenance of epithelial differentiation.
Forkhead proteins HNF3α, -β, and -γ (Costa et al., 1989; Lai et al., 1990) interact with the corresponding sites as monomers (Clark et al., 1993). A significant structure similarity of HNF3 "winged helix" DNA binding domain to histones provides these factors with the ability to bind the compacted chromatin and alter its nucleosomal organization during gene activation (McPherson et al., 1993). This property has been proposed to play the decisive role in hepatogenesis. Embryonic specification of liver is determined by signals from the cardiac mesoderm cells inducing alterations in gene expression and morphology of endodermal epithelial cells (for review see Zaret, 2002). The competence of these cells to enter hepatic differentiation is attributed to the activities of HNF3 and GATA transcription factors recognizing the corresponding binding sites and opening the compacted chromatin structure of hepatic genes (Cirillo et al., 2002).
During embryonic development HNF3β, -α, and -γ are activated successively in the definitive endoderm with HNF3β being detected at the onset of gastrulation (Ang et al., 1993; Monaghan et al., 1993). Even HNF3β inactivation leads to early embryonic lethality associated with abnormal development of the foregut endoderm that gives rise to the liver and pancreas (Ang and Rossant, 1994; Weinstein et al., 1994; Dufort et al., 1998). Targeted disruption of HNF3α gene causes postnatal growth retardation and death within the first week of life due to glucose homeostasis failure (Kaestner et al., 1999). Mice with inactivated HNF3γ have no developmental abnormalities and differ from the normal animals by altered transcription of several liver-specific genes (Kaestner et al., 1998). Based on these data, HNF3β has been proposed to play the decisive role in early hepatic specification.
Note that HNF3β inactivation in the adult liver does not entail any significant changes in hepatic function and morphology (Sund et al., 2000), suggesting essential alterations in HNFTs function and regulation at different stages of development.
HNF4α is nuclear hormone receptor (Sladek et al., 1990) which binds DNA exclusively as homodimer (Jiang et al., 1997). HNF4α is the main activator of HNF1α expression which in turn regulates a wide range of liver-specific genes (Kuo et al., 1992). Also, HNF4α directly regulates the transcription of numerous genes essential for hepatocyte differentiation and function.
Two groups of HNF4α isoforms originating by different promoter usage have so far been identified (summarized in Sladek and Seidel, 2001). The variants transcribed from P1 promoter are predominant in the adult liver and differentiated hepatomas (Nakhei et al., 1998; Torres-Padilla et al., 2001). The isoforms regulated by an alternative promoter P2 are mainly expressed in stem cells, embryonic liver, pancreatic β-cells, and dedifferentiated hepatoma cell lines (Nakhei et al., 1998; Thomas et al., 2001; Torres-Padilla et al., 2001). Two groups of isoforms have different impacts on target genes expression (Torres-Padilla et al., 2001) apparently due to different patterns of interaction with coactivators (Torres-Padilla et al., 2002). The "embryonic" forms more effectively activate the promoters of early hepatic genes AFP and transthyretin, while "adult" variants have a more significant impact on the transcription of mature hepatic markers (Torres-Padilla et al., 2001). HNF4α transcription from P1 and P2 promoters is driven by distinct mechanisms and the ratio of resulting two groups of isoforms (referred hereafter as α1 and α7) influence the maintenance of hepatic differentiation.
During mouse embryonic development HNF4α mRNA is first identified on day 4.5 in the primitive endoderm (Taraviras et al., 1994; Chen et al., 1994). Inactivation of the HNF4α gene leads to embryonic lethality at day 10 due to a block in visceral endoderm differentiation (Chen et al., 1994; Duncan et al., 1997). This defect can be rescued by complementation of HNF4α -/- embryos with a tetraploid embryo-derived visceral endoderm (Duncan et al., 1997). While HNF4α appeared to be dispensable for early specification of hepatic lineage, it was found to be essential for the subsequent steps of hepatic differentiation and for metabolic regulation and liver function (Li et al., 2000). Loss of HNF4α expression in midgestation induces dramatic abnormalities of liver morphology linked with disruption of cell adhesion and cell junction contacts and downregulation of the number of genes critical for epithelial morphology maintenance (Parviz et al., 2003). Conditional knockout of HNF4α in the adult liver suggests that this factor is the central regulator of genes involved in lipid and urea homeostasis (Hayhurst et al., 2001; Inoue et al., 2002). Thus, the recent experiments on knockout mice have clearly demonstrated the essential role of HNF4α in liver differentiation and morphogenesis at different stages of development.
HNF6 binding the same sites as HNF3 is expressed in the adult liver and pancreas (Samadani and Costa, 1996; Lemaigre et al., 1996).
In the liver, HNF6 is expressed both in hepatocytes and in cholangiocytes from the initial steps of their development. The HNF6 knockout in mice causes the abnormal development of intra- and extrahepatic bile ducts, absence of gallbladder, hepatic artery malformations, and cholestasis (Clotman et al., 2002; Clotman et al., 2003). Defects in the development of the biliary tract are similar to those observed in liver-specific HNF1β -/- mice (Coffinier et al., 2002; Clotman et al., 2003). Moreover, HNF6 was shown to control the expression of HNF1β in the embryonic biliary epithelial cells (Clotman et al., 2002). HNF6 inactivation is also associated with pancreatic abnormalities and diabetes mellitus due to defects in endocrine cells differentiation (Jacquemin et al., 2000). These data suggests that HNF6 is a key regulator of hepatoblasts differentiation into biliary epithelial cells and endocrine lineage differentiation in pancreas.
C/EBP (CCAAT/enhancer binding) proteins comprise the most widespread family of transcriptional regulators binding CCAAT-box as homo- or heterodimers (Landschulz et al., 1988; reviewed in Ramji and Foka, 2002; Schrem et al., 2004). Being expressed in different combinations in a wide range of tissues and cell types, C/EBP proteins regulate the variety of essential physiological processes like organogenesis, differentiation, apoptosis, inflammatory response, metabolism and some others. Transcriptional activity of C/EBP family members is controlled tissue- and stage-specifically at multiple levels, and is responsive to hormonal, mitogenic, nutrition and stress signals (reviewed in Ramji and Foka, 2002).
C/EBPα and β genes predominantly expressed in the liver produce multiple protein isoforms with different trans-activation properties (Descombes and Schibler, 1991; Ossipov, 1993). Specifically, one of C/EBPβ isoforms, liver-enriched transcriptional inhibitory protein (LIP), lacks the functional trans-activation domain but preserves the ability for dimerization with other family members, acting as a dominant negative regulator of C/EBP dependent transcription (Descombes and Schibler, 1991). The multiple control of C/EBP transcriptional activities does ensure the strict regulation of their impact in diverse biological processes.
In the liver C/EBP factors regulate the expression of functional hepatic genes and mediate the acute phase response. Moreover, C/EBPα is associated with terminal differentiation and inhibits hepatocyte proliferation, while C/EBPβ exerts the opposite effect. The balance of C/EBPα and β reflects the proliferative state of hepatocytes: during regeneration, the level of C/EBPβ significantly increases while that of C/EBPα rapidly falls (Greenbaum et al., 1995; Rana et al., 1995). C/EBPα transcription is strongly enhanced after the birth when hepatocytes differentiate and convert to the quiescent state (Wang et al., 1995).
Mice lacking C/EBPα die shortly after birth because of impaired glycogen synthesis and storage (Wang et al., 1995). These animals have defects in lung development, increased hepatic proliferation, and distortion of liver architecture with acinar formation (Flodby et al., 1996). The primary hepatocyte cultures obtained from C/EBPα -/- neonatal livers exhibit rapid growth, chromosomal instability, and increased transformation frequency, but maintain the hepatocyte-like morphology with cell-cell contacts and albumin expression (Soriano et al, 1998).
Mice with inactivated C/EBPβ are viable but display defective differentiation of adipose, ovarian and mammary gland (see Ramji and Foka, 2002 and references therein). C/EBPβ -/- mice demonstrate an impaired hepatocyte proliferation in response to partial hepatectomy (Greenbaum et al., 1998).
Note that C/EBPβ is able to induce pancreas to hepatic trans-differentiation. Enforced expression of C/EBPβ in a pancreatic cell line, AR42J-B13, causes downregulation of pancreatic marker amylase, translocation of HNF4α to nucleus, and activation of a set of liver-specific genes (Shen et al., 2000). This example shows that differentiation and functional program of epithelial cells can be critically modified by altered activities of very few tissue-specific transcriptional regulators.
The transcriptional pattern of each HNF is not restricted to the liver alone but all transcription factors of that class are expressed only in this organ. The impact of each transcription factor on liver differentiation is not static and significantly alters in the course of development (Jochheim et al., 2004). The transcriptional hierarchy of different HNFs is highly complex (reviewed in Cereghini, 1996; Lazarevich, 2000; Locker, 2001) and not completely solved yet.
There is growing evidence that the balance of HNFs determines to a great extent the fate of different cell lineages in organogenesis. All the critical steps of liver formation are preceded or accompanied by modulation of HNFTs network. It is likely that HNFs are responsible for the competence to inductive signals coming from different mesodermal cell types (reviewed in Zaret, 2002; Lemaigre and Zaret, 2004) and reprogramming of gene transcription induced by these signals. Sequential activation of HNF3β, HNF6 and HNF4α accompanies in vitro differentiation of murine embryonic stem cells towards hepatic phenotype (Jochheim et al., 2004).
HNF3 factors are essential for acquisition of the endoderm competency to adopt a hepatic fate (Zaret, 2002). HNF1β and HNF6 mediate hepatoblasts differentiation into cholangiocytes and bile ducts formation (Clotman et al., 2002; Coffinier et al., 2002). HNF4α (Parviz et al., 2003) and C/EBPα (Wang et al., 1995) are essential for differentiation of embryonic hepatocytes and HNF1α plays an important role in the functional maturation of hepatocytes during the postnatal period (Pontoglio et al., 1996; Akiyama et al., 2000).
Thus, specification of hepatocytes and cholangiocytes, endodermal components of the liver originated from the common progenitors, depends upon differential expression of HNF4α, HNF1α, C/EBPα and HNF6, HNF1β respectively. Importantly, reversible dedifferentiation of normal hepatocytes in primary culture is accompanied by decrease in activity of HNF4α, HNF1α, C/EBPα and activation of HNF3 proteins indicating that these factors can mediate the alteration of hepatocyte transcriptional program in response to disturbing of normal liver structure (Section III.F).
In summary, having been primarily identified as transcriptional regulators of functional hepato-specific genes, HNFs were then clearly shown to influence a variety of cell and tissue characteristics, particularly proliferation, morphology, organogenesis, apoptosis, stress response, etc. The wide range of processes modulated by this class of factors and the profile of HNFs expression suggests their involvement in similar regulatory processes taking place in other epithelial structures.
2. HNFs in non-hepatic epithelia
While HNFs were first discovered in the liver, they are in different combinations expressed in the epithelia of other organs like pancreas, kidney, lung, intestine, stomach, mammary gland, skin (reviewed in Tronche and Yaniv, 1992, Cereghini, 1996; Lazarevich, 2000; Locker, 2001), and clearly participate in gene regulation and tissue differentiation. The tissue-specific regulatory network in these organs is formed by cooperative action of HNFs and additional transcriptional regulators specific for definite cell lineage. Below, we will focus on the place of HNFs in the regulatory cascade that defines the development of pancreas and then briefly summarize the impact of liver-enriched transcriptional factors on the differentiation of other organs.
Being derived from the common ontogenetic precursor, the primitive foregut, the liver and pancreas demonstrate very similar patterns of HNFs expression (Locker, 2001). Notably, the most significant difference in HNF set between the liver and pancreas are substitution of "adult" isoforms of HNF4α by "embryonic" ones and expression of HNF4γ absent in hepatocytes. Activation of the P2 promoter, driving the expression of embryonic isoforms of HNF4α, can be attributed to the activity of Pdx1 and HNF1 factors which bind the P2 promoter (Thomas et al., 2001), and to the absence of adult HNF4α isoforms reported to suppress α7 promoter activity (Briancon et al., 2004). Perhaps these differences are not sufficient to define the divergence of the developmental fate of the liver and pancreas; moreover, the regulatory links revealed in the liver transcriptional network are altered in pancreas by the addition of new players. Still, the significance of HNFs in pancreas development is apparent.
Pancreas arises from the fusion of the ventral and dorsal buds of primitive gut epithelium. It undergoes a complex set of morphological transitions giving rise to several highly specialized cellular lineages (summarized in Slack, 1995; Kim and Hebrok, 2001; Habener et al., 2005). The mature pancreas consists of two functional compartments. Exocrine acinar and duct cells are responsible for secreting enzymes into the digestive tract. Endocrine hormone-secreting cells are located in the islets of Langerhans. This population includes four functionally specialized cell types: insulin-producing β-cells, glucagon-producing α-cells, somatostatin-producing δ-cells, and pancreatic polypeptide-producing PP-cells. Specification and differentiation of this complex structure are driven by signaling from different patterns of the overlying mesoderm and are accompanied by induction of a branching cascade of transcription factors.
The early stages of pancreatic development are defined by the activity of homeodomain and basic helix-loop-helix factors Pdx1, Ptf1a, Hlxb9, Isl1, or Hex, while additional set of transcriptional regulators, namely, Neurogenin-3, Pax4, Pax6, NeuroD/β2, and Nkx2.2, is essential for pancreatic lineage specification and function (Edlund, 2001; Habener et al., 2005). HNFs widely cooperate with these factors in pancreatic organogenesis and lineage specification. Liver-enriched transcription factors were found to regulate transcription of Pdx1, Pax4, Neurogenin-3, and Nkx2.2 genes. Recently, Cereghini laboratory has reported that HNF1β deficiency in mice causes pancreas agenesis due to the failure of ventral pancreas bud specification and reduction of the dorsal pancreas, caused by diminished cell proliferation (Haumaitre et al., 2005). HNF1β inactivation induced a dramatic deregulation of the pancreatic transcriptional network including the repression of transcription factor Ptf1a, essential for the acquisition of pancreatic fate, loss of early endocrine pancreatic markers, and ectopic expression of Sonic Hedgehog and Indian Hedgehog signaling molecules, which regulate regional specification of embryonic gut endoderm. These complex abnormalities result in defective developmental patterning of the primitive gut. These findings allow to place HNF1β to one of the top positions of the currently identified hierarchy of transcription factors defining early pancreatic morphogenesis.
Importantly, transcription factors may influence pancreatic differentiation not only by direct regulation of transcriptional programs but also morphogenetically, as was recently shown in Zaret laboratory for the homeobox gene Hex, required for the onset of hepatogenesis (Bort et al., 2004). Hex regulates proliferation of cells at the leading edge of the ventral endoderm and thus their positioning relative to the cardiogenic mesoderm, which can trigger the pancreatic differentiation program. It is likely that similar mechanisms also mediate the morphogenic properties of some other transcriptional regulators.
HNFs play a key role in the function of mature pancreatic β-cells, particularly, in regulation of insulin secretion. While HNF1 family members can regulate insulin gene activity by direct binding to the promoter sites, HNF4α, HNF3β and HNF6 likely affect insulin transcription indirectly through modulation of HNF1, Pdx, or NeuroD/β2 expression (Habener et al., 2005). Mutations of the human HNF4α, HNF1α or HNF1β genes cause an autosomal dominant form of non-insulin-dependent diabetes mellitus called maturity-onset diabetes of the young (MODY), type 1, 3, and 5 respectively, while types 4 and 6 are attributed to Pdx1 and NeuroD/β2 mutations (reviewed in Ryffel, 2001; Habener et al., 2005).
Studies on animal models confirm the importance of HNF1α, HNF1β and HNF4α for pancreatic β-cell differentiation (Pontoglio et al., 1998; Wang et al., 2004; Haumaitre et al., 2005; Gupta et al., 2005). An abnormal pancreatic phenotype with perturbed differentiation of endocrine cells was also revealed in HNF6 -/- mice (Jacquemin et al., 2000). Disruption of HNF3β in pancreatic β-cells results in deregulation of insulin and glucagon secretion (Sund et al., 2001). Importantly, HNF3 family members are also involved in pancreatic α-cells differentiation and regulation of proglucagon transcription (Kaestner et al., 1999; Lee et al., 2005).
The disturbance of normal HNF function may influence not only functional but also morphological properties of pancreatic cells. For example, expression of a dominant-negative form of human HNF1α in pancreatic β-cells (Yamagata et al., 2002) impaired both insulin secretion and tissue morphology, which was due to the disruption of E-cadherin-dependent cell adhesion in the islets. The impairment of HNF1 function also diminished the expression of E-cadherin. The islet structural abnormalities were similar to those observed in transgenic mice expressing dominant-negative E-cadherin (Dahl et al., 1996). Taking into account that due to dimerization with endogenously expressed forms a dominant-negative HNF1α can block the function of both family members, these findings indicate that E-cadherin gene is a potential target for HNF1-responsive regulation in the pancreas.
Members of HNF1 family are both expressed in the kidney, but exhibit distinct patterns of expression (Lazarro et al., 1992; Pontoglio et al., 1996). Apparently, these factors are essential for kidney morphogenesis and regulate the number of genes defining renal function and morphology. Importantly, inherited mutations of HNF1α (MODY3) cause reduced tubular reabsorption of glucose (Pontoglio et al., 2000), while HNF1β mutations (MODY5) are associated with renal cystic abnormalities and/or genitourinary defects (Nishigori et al., 1998). HNF1α knockout induced the renal Fanconi syndrome characterized by urinary glucose loss in mice (Pontoglio et al., 1996). HNF1α was shown to regulate proximal tubule-specific gene expression, while HNF1β regulates the number of genes responsible for renal cystogenesis, and kidney-specific Ksp-cadherin (Igarashi, 2003; Gresh et al., 2004). Renal-specific inactivation of HNF1β causes the derangement of kidney tubular differentiation and renal cystic abnormalities (Gresh et al., 2004).
Members of the HNF3 family were also proposed to mediate epithelial-mesenchymal interactions in embryogenesis. Together with thyroid transcription factor 1 and other forkhead proteins, HNF3α and HNF3β regulate signaling and transcriptional programs required for morphogenesis and cell differentiation during formation of the lung (Costa et al., 2001). As indicated by mice knockout studies, HNF3α and HNF3β control cell proliferation, regulate transcription of lung epithelial cell markers, and control branching morphogenesis, possibly, through regulation of Sonic hedgehog transcription (Wan et al., 2005). HNF3α was also reported to regulate prostate-specific expression of PSA gene (Gao et al., 2003).
In concordance with their wide tissue distribution, C/EBP proteins are involved in differentiation and/or proliferation control in a variety of organs including skin, intestine, lung, adipose tissue, and mammary gland.
For example, C/EBPα, -β and -δ are differentially expressed throughout the mammary gland postnatal development. The differentiation stages of the mammary gland during pregnancy and lactation are clearly associated with regulating the balance between differentiation, proliferation, and apoptosis (Hennighausen and Robinson, 1998). C/EBPβ is essential for normal growth and functional differentiation of the mammary gland (Seagroves et al., 1998; Robinson et al., 1998). C/EBPβ-deficient mammary epithelium was shown to be defective in both proliferation and differentiation during pregnancy and failed to express the milk protein genes. In transplantation experiments, the C/EBPβ -/- mammary gland fat pad was able to support the normal development of wild type epithelial cells, which indicates that this effect is not influenced by C/EBPβ activity in the stromal component. Thus, the abnormal development of mammary gland in the absence of functional C/EBPβ is a likely consequence of disturbed hormonal regulation or other extracellular signaling cascades in mammary gland epithelial cells.
C/EBPα and β have discrete expression patterns in epidermis and are implicated in keratinocyte differentiation (Zhu et al., 1999). In accordance with previously pointed role of CEBPα as a terminal differentiation factor, it is involved in lineage-specific maturation in lung (Flodby et al., 1996) and intestinal epithelium (Chandrasekaran and Gordon, 1993). C/EBP family members are also involved in the regulation of tissue-specific gene expression and in the acute phase response in certain epithelial cell types.
Taking into account that the early development of certain epithelial organs, like the pancreas, lung or intestine, is defined by strict continuity of growth factor signaling, cell-cell and cell-ECM interactions, it is not surprising that some liver-enriched factors, which transduce these signals into alterations of the transcriptional program during liver organogenesis, conserve similar function in other endodermal lineages. Importantly, transcription factors may realize the developmental programs not only by direct regulation of transcription but also by modulation of proliferative and morphological properties of cells, which in turn define their proper positioning during tissue specification in the embryo (Bort et al., 2004). Although the impact of liver-enriched factors on cell differentiation is essentially modified by cooperation with other tissue-specific regulators, some basic principles of HNFs influence on physiologic properties, proliferation, and maintenance of epithelial morphology remain conserved.
III. Microenvironmental control of transformed cells
A. Control of transformed cells by normal surrounding
It is well known that the epithelial cell layer separating different tissues from each other and from extracellular fluids contains several kinds of impermeable contacts, both for cells and soluble macromolecules – intercellular adhesion contacts. They include different types of junctional contacts, such as tight junctions, gap junctions, and intercellular contacts through adhesion molecules, as well as contacts with ECM by means of integrin receptors. These contacts determine the epithelial cell polarity, integrate cells into uniform tissue, and participate in their functioning.
On their way to overt neoplasms, transformed cells should be liberated from limitations overlaid by the surrounding tissue, i.e. from influence of intercellular and cell-matrix contacts, including interaction with basal membrane.
There is no doubt that epithelial tumors have some "memories" about the differentiated state of their progenitor cells. For example, primary liver tumors induced by a "mild" carcinogen always retain tissue-specific antigens, at least some of them (Abelev, 1965; Guelstein and Khramkova, 1965; Khramkova and Guelstein, 1965). Primary and even subcutaneously transplanted carcinogen-induced hepatomas look like pieces of liver after staining with liver-specific antibodies (Engelhardt et al., 2000). However, long-term transplanted hepatomas lose a significant part of tissue specific antigens in the course of progression.
What barriers should be overcome, at least at the initial steps of progression? The first barrier is the control of microenvironment exerted by the normal tissue. That was formulated by Weinberg (1989), while the first experimental evidence for this statement was offered much earlier by demonstration of initiation and promotion in carcinogen action (Berenblum, 1954). Initiation is induced by a subcarcinogenic dose of chemical carcinogen, while promotion "develops" the initiated tissue by noncarcinogenic substances. Initiation is maintained for long time and initiated cells are able to produce neoplasms within months and years after initiation. Initiation is similar to transformation or to some critical step leading to transformation, while promotion leads to the development of overt neoplasm from initiated cells. Initiation could be a transforming mutation while promotion is apparently a condition favoring selection of a transformed clone from targeted cells. One of the strongest promoters, phorbol ester, TPA, stimulates anchorage independent growth of an initiated clone (Dotto et al., 1985; Karen et al., 1999). This property is typical for growth of malignant clones, rather than normal cells. Promoters have many different activities but among them there is a property common for various promoters, i.e. block of gap junctional communications (Yamasaki et al.; 1990; Murray and Fitzgerald, 1979; Krutovskikh, 2002).
Weinberg (1989) demonstrated that individual ras-transformed cells dispersed with low frequency in a monolayer of normal cells were effectively controlled by the latter ones. A massive infection of cells with oncogenic virus bearing the same oncogene (ras) developed regular transformation focuses.
Mixture of transformed and normal cells forms tumor nodules in vivo only when transformed cells highly predominate.
This means that there is a universal system controlling the behavior of individual cells in the tissues (metabolic cooperation). It is unknown how such control is performed, but there are reasons to believe that disruption of gap junctional communications leads to impairment of this control. All these data suggest that transformation and malignancy could be separated in carcinogenesis and that malignant properties arise during progression of initially transformed cells.
A new step along this way has been recently made by Laconi and his colleagues. They found that normal or initially carcinogen-transformed rat hepatocytes, injected intravenously to normal partially hepatectomized rats, were integrated into the structure of regenerating normal liver without signs of tumor growth. However, poisoning of recipient animals with retrorsine, alkaloid which inhibits hepatocyte division, led to 80-90% replacement of recipient liver with donor cells (Laconi et al., 2001a). Transplantation of carcinogen-initiated liver cells into retrorsine-treated rats resulted in development of overt neoplasms during the process of recipient liver replacement (Laconi et al., 2001b; Dabeva et al. 2002).
The above data once again confirmed the existence of normal cellular barrier on the way of initiated cells to malignancy, but the nature of this barrier is still unknown.
And there is one more system, which allowed not only to demonstrate the "normal" control of tumor progression but also to disclose a little bit the underlying events. The system included a 3D model of human skin keratinocytes seeded on basal membrane covered with skin fibroblasts. Intraepithelial nodules consisting of premalignant (initiated) keratinocytes could be reversed into normal keratinocytes when seeded in large excess of normal cells (25:1), but at 4:1 ratio the cell culture behaved as an initial step of tumor progression. It was promoted by basal membrane which stimulated outgrowth of progressed keratinocytes and created a significantly increased tumor-promoting effect (Javaherian et al., 1998; Vaccariello et al., 1999; Andriani et al, 2003).
Connective tissue elements associated with keratinocyte outgrowth became components of a future stroma, whose role is decisive at least at the early stage of tumor progression. The role of stroma in tumor development changes in the process of progression from induction of a tumor clone due to independence from growth factors to its invasive growth, loss of tissue architecture, and, finally, to its metastatic growth (Cunha et al., 2003).
Increasing understanding of the role of stroma in tumor development is reflected in a number of recent analytical reviews (De Wever and Mareel, 2000; Pupa et al., 2002; Cunha et al., 2003).
Thus, there is no doubt that "initiated" cells should overcome restraining limitations of the normal cellular microenvironment, i.e. interrelations with normal neighbours (Radisky and Bissell, 2004). Such "initiated" cells were found not only in carcinogen-treated but also in the normal breast tissue (Holst et al., 2003).
We know only some elements of these interactions and gap junctions are among them. It is known that disruption of these communications (metabolic cooperation) is always associated with the promoter action (Yamasaki, 1990; Krutovskikh, 2002) and is, probably, necessary, at least for maintaining the epitheliocyte differentiation state. Hence, this first step of progression is most probably associated with the initial steps of epithelia dedifferentiation. Further steps are also associated with continuing progressive loss of epithelial differentiation.
B. From intercellular interactions to interaction with ECM
Analysis of a differentiation marker of hepatocellular carcinoma (HCC), AFP, led us to the establishment of a crucial role of intercellular relations in liver cell differentiation and then to the most important factor of those relations – cell-ECM interactions.
AFP is expressed in the yolk sac endoderm, fetal liver, and, transiently, in the regenerating liver. AFP synthesis is resumed in HCC (Abelev, 1971; Abelev and Eraiser, 1999).
Immunohistochemical analysis of CCl4-induced liver regeneration in mice revealed a clear-cut localization of AFP, strictly in the perinecrotic area of damaged liver, more precisely in one layer of cells surrounding this area (Engelhardt et al., 1984; Gleiberman and Abelev, 1985). Detailed immunohistochemical analysis of cells in the perinecrotic layer revealed that these cells gradually lost their polarity with the disappearance of apical bile canaliculi antigen, Bgp1 (Kuprina et al., 1990), and behaved like cells isolated from the liver plate structure. Simultaneously they re-express AFP.
Immunohistochemical study of AFP fading in early ontogenesis showed that the decrease of AFP expression is gradient-like starting in the portal area and ending around the central veins (see Gleiberman and Abelev, 1985; Spear, 1999).
The "rings" around the central veins are the latest sites of expression AFP before the complete disappearance of this protein. This process coincided with the liver plate formation, which starts in the region of portal tract and spreads toward the central veins. The appearance of apical marker Bgp1 and its concentration at the bile capillaries is an excellent marker of hepatocytes in the liver plate, where it borders bile capillaries. Thus, AFP synthesis suppression is associated with hepatocytes incorporation into the structure of liver plate. Drop out of liver plate leads to re-expression of AFP, as clearly seen in the liver during regeneration after treatment with different hepatotoxins (Kuprina et al., 1985). The association of AFP suppression with the liver plate formation was defined as "structural repression" (Abelev 1978) or "architectural regulation" (Notenboom et al., 1996) – the terms stressing the association of AFP suppression with cell-cell interactions in morphogenetic process. In the latter work, a suspension of fetal rat hepatocytes was introduced into the spleen of young recipient rats. When transplanted cells build liver plate structures, they cease to synthesize AFP. This process was associated with establishment of the lobular pattern of liver-specific enzymes expression and AFP transcription (Moorman et al., 1990; Notenboom et al., 1996).
Direct evidence of the significance of cell-cell contacts for hepatocyte differentiation was obtained in experiments with a suspension of isolated mature hepatocytes. Perfusion of the liver with collagenase led to dissociation of the liver tissue into a suspension of single hepatocytes. Transfer of the hepatocyte suspension onto plastic dishes led to adhesion of the cells to plastic, where they formed a sparse monolayer without intercellular junction communications. Individual cells in the monolayer lost their polarity and Bgp1 antigen. In a few days, they began to synthesize AFP very actively. They showed dedifferentiated phenotype, typical to the hepatocytes liberated out of plate structure. (The distribution of HNFs, typical for dedifferentiated hepatocytes is described in II B). As a result of gentle rotation of culture dishes, cells were gathered around the center, and dense center and sparse periphery were formed. AFP synthesis was visualized as a ring only in the periphery, and gap junction communications were seen only in AFP-negative dense central part of the monolayer (Gleiberman et al., 1989a). Thus, clear association of gap junctions with AFP suppression, which served as a "marker" of mature hepatocyte, took place.
But it was only a part of the whole picture. According to earlier data on the role of ECM in maintaining hepatocyte shape and function (Guguen-Guillouzo et al, 1983; Ben Zeev et al., 1988) the effect of 3D ECM on differentiation markers synthesis was investigated.
When hepatocytes were placed on collagen (type I) dried in the plastic dish, they formed a sparse monolayer similar to that formed on plastic alone. But if cells were inserted between two layers of fresh collagen gel they formed liver-like islands of highly organized hepatocytes. Cells in these islands were cuboidal and polygonal, connected by gap junction communications, forming bile canaliculi, and expressing Bgp1 antigen at the border of bile capillaries. The cells produced serum albumin, while AFP synthesis was completely suppressed, i.e. they were typical mature hepatocytes (Gleiberman et al., 1989b). Thus, 3D ECM was necessary for maintenance of the mature type differentiation of hepatocytes. This effect required the presence of 3D ECM but was independent from the collagen nature: type I instead of type IV present in the liver.
The same effect was obtained in a mixed culture of hepatocytes with non-parenchymal liver cells as was earlier suggested by Guguen-Guillouzo et al. (1983) and reproduced later by Gleiberman et al. (1989b). In this system, not only gap junction communications were clearly seen, but abundant 3D ECM was produced. Under such conditions, hepatocytes performed their physiological functions, i.e. drug detoxication and serum protein synthesis. Principally similar results were obtained by Di Persio et al. (1991) with serum albumin synthesis by hepatocytes. Those authors showed that serum albumin enhancer was activated significantly in 3D ECM, which determined cuboidal shape of hepatocytes and their differentiation.
Thus, these experiments were among the first indications to the role of 3D ECM in epithelial differentiation.
Recent study of Kudrjavtseva and Engelhardt (2003) showed that Ras-transformed variant of IAR (non-parenchymal liver cell line) in a mixed culture with hepatocytes produced defective ECM, which did nor suppress AFP synthesis and did not support the liver-like cellular organization. The same result was observed with RSV-transformed IAR cells. Normal IAR in control experiments supported hepatocyte maturation with AFP-suppression.
These observations have something in common with the recent data demonstrating that radical stromal changes or even stromal cell transformation are necessary for epithelial cell transformation leading to prostate carcinoma (Cunha et al., 2003) or breast carcinoma (Bissell et al, 2002; Kuperwasser et al., 2000).
Thus, cell-cell communications and cell-matrix interactions appeared to be necessary for the initiation of hepatocyte differentiation or at least for its maintenance. It was shown that embryonic hepatocytes more actively express embryonic liver enzymes while growing on embryonic liver basal membrane ECM than on adult liver basal membrane ECM and vice versa (Brill et al., 2001).
Now we would show that the regularities found in liver development are of general significance for the epithelial organs. We will discuss the mammary gland formation and functioning. An attempt to construct a mouse model for human mammary gland studies was made by transplantation of human breast epitheliocytes into nude or scid mice. But transplantation failed unless the human fat pad and stromal fibroblasts were preliminarily transplanted. Only transplantation of human breast epithelium together with tissue (or cells) creating the mammary gland microenvironment led to the formation of functioning (casein synthesizing) mammary gland in mice (Kupewasser et al., 2004).
Normal differentiation of mammary gland epithelium, as was shown on the breast tumor tissue, was restored by interaction of integrin system of this tissue with the blocking antibody to β1-integrin in 3D ECM (Weaver et al., 1997; Chrenek et al., 2001). These data, though obtained on tumor models, clearly show that at least maintaining mammary gland differentiation requires 3D ECM and is realized through integrins (see also Weaver et al., 2002).
Very interesting experiments on cell-ECM relations were carried out by Cukierman and her collaborators with the use of fibroblasts (Cukierman et al., 2001; 2002; Yamada et al., 2003). They used the elongated shape of fibroblasts and their ability to synthesize collagen and fibronectin, as criteria of fibroblast differentiation. These properties of mature fibroblasts were observed only in 3D collagen gel of homologous ECM. The same ECM pressed to 2D plate did not induce fibroblast maturation, like heterologous ECM (Cukierman et al., 2001). These data emphasized the importance of ECM origin (chemical composition?) as well as its 3D structure.
Cell-ECM interactions are realized by integrins which specifically recognize ECM molecules and influence cell microenvironment "from inside" the cell (Hynes, 2002; Danen and Sonnenberg, 2003).
Wide variability of integrins composition and their conformational liability enable these molecules to recognize different components of ECM, as well as to transduce different signals, produced by ECM alone or in association with growth factors, into cells (Yamada et al., 2003; Faraldo et al., 2002).
All those indications to the importance of 3D ECM for cell differentiation raised a problem: how does spatial arrangement of cell-matrix interaction lead to activation of tissue specific genes? There are only few attempts to understand the mechanisms underlying these interactions, when it was shown that nuclear shape and nuclear matrix configuration depended on the 3D structure of ECM (Lelievre et al., 1998).
Anyway, it is clear that rearrangement or disruption of such interactions taking place in tumor progression lead to derangement or loss of differentiation in epithelial tumors.
C. Dedifferentiation in carcinomas as consequence of tumor progression
This paper considers transformation and progression as distinct events in the origin and evolution of malignant growth. As was shown above, transformation is a process not necessarily associated with the loss of differentiation state of tumor progenitor cell. It can be blocked, or "frozen", at certain stages of differentiation, with the inhibition of later stages, but with retaining of the preceding ones (Potter, 1978; Greaves, 1982; Abelev, 2000; Tenen, 2003). These properties are typical especially for hemoblastoses and are routinely used for their classification based on immunophenotyping. Rare cases of "mixed" phenotype, typical for different hemopoietic cell lines, are usually observed in acute leukemia, derived from the earliest stages of progenitor cell differentiation, expressing markers of various lines of differentiation (Greaves, 1986; Van Dongen et al., 2002; Kenting et al., 2002).
Highly differentiated (minimal) Morris hepatomas illustrate carcinomas, often demonstrating high degrees of differentiation, as well as antigen retaining in mouse hepatomas (Section III.A). Morphological similarity of epithelial tumors to tumor progenitor tissue is one of the features widely used in pathomorphological tumor diagnosis.
Intermediate filaments (Ku et al., 1999) of epithelial tissue (cytokeratins) are the most conservative markers of different types of epithelia and are widely used in carcinoma classification and diagnostics, as well as in micrometastases detection (Braun et al., 2000).
So called tumor markers, such as AFP, CEA, prostate specific antigen (PSA) and CA-125 (ovarian cancer serum marker), are tissue specific antigens, specific to fetal or adult stages of certain tissues development (reviewed in Abelev and Sell, 1999).
Thus, a more or less differentiated status is maintained in epithelial tumors but it changes in the process of tumor progression, most probably due to altered tumor cell interactions with the microenvironment.
1. Loss of gap junctional communications
The first barrier, which should be overcome in the course of tumor progression, is the normalizing effect of surrounding normal cells. It has been extensively studied with chemical carcinogens, which allowed discrimination between initiation and promotion. Promoter effect leads, among others, to gap junctional communication disruption or blockage, as we discussed earlier (Section III.A).
It is not yet clear how this blockage induce the loss of differentiation but it does lead to it since down-regulation of differentiation in sparse monolayer of isolated hepatocytes is always associated with a block of gap junctional communications (Section III.B). Conversely, induction of gap junctional communication by ECM enhances mammary epithelial cell differentiation marked by beta-casein upregulation (El-Sabban et al., 2003).
2. Cadherins and adherence molecules
The next barrier includes cell junctions through cadherins and adherence molecules. In the epithelia, the most abundant member cadherin family is E-cadherin, which is responsible for immediate contact of neighbouring cells (Cavallero and Christofori, 2004; Peinado et al., 2004, Section II.A). E-cadherin is the central component of intercellular contacts: it connects the network of intermediate filaments of different cells with each other and enters the occlusion bodies. Hence, it creates a continuous network between homologous cells and integrates individual cells into the entire tissue (Cavallero and Christofori, 2004). Downregulation of E-cadherin leads to tissue dissociation into separate cells and is a general component of tumor progression. Conversely, transfection of E-cadherin gene stops tumor progression and restores the ability of dissociated cells to form tissue-like aggregates (see Christofori and Semb, 1999; Arias, 2001; Thiery, 2002). E-cadherin has not only structural functions, but is linked to several signaling pathways (Matsumura et al., 2001; Behrens et al., 1993). The cytoplasmic domain of E-cadherin is associated with α, β and γ-catenins, while β-catenin can act as nuclear transcription factor responsible for regulation of proliferation and differentiation. E-cadherin gene expression may be affected by mutation, methylation or repressed by transcription factor, particularly Snail or Slug (see Peinado et al., 2004). When E-cadherin gene is downregulated or disrupted, β-catenin is liberated, migrates into the nucleus, and stimulates proliferation and tissue-specific gene transcription.
One of the main signaling cascades, Wnt, participating in carcinogenesis is linked to β-catenin, which can modify the activity of this pathway (Cavallero and Christofori, 2004).
Both functions of E-cadherin, structural and signaling, are associated with cell differentiation, i.e. expression of tissue-specific genes, but precise ways of realization of this function are not yet clear.
Impermeability of epithelial cell layer for migrating cells and for fluids is due mainly to E-cadherin presence. Downregulation or loss of E-cadherin is a very characteristic step in epithelial tumor progression (Perl et al., 1998; Strathdee, 2002). One of the consequences of E-cadherin downregulation is disruption of gap junctional communications, which are necessary for normal cell functioning (Krutovskikh, 2002). It is an additional step in dedifferentiation associated with tumor progression.
3. Integrins – ECM relations in tumor progression
Tissue-specific integrins are partially lost during progression of many epithelial tumors. In parallel, these tumors lose the ability to recognize matrix components, i.e. fibronectin, the main component of ECM (Ruoslahti, 1999). Loss of recognition of ECM is probably accompanied by the gain of anchorage independent proliferation, which is the most characteristic feature of tumor cells.
Role of integrin derangement in tumor progression seems to be very important, since integrins specifically and variably recognize amorphous ECM, as well as basal membrane. Partial redifferentiation or reversion of epithelial tumors through specific action on their integrin system is the evidence of the impact of integrin downregulation or derangement in the dedifferentiation of corresponding epithelial tumors (Kenny and Bissell, 2003; Danen and Sonnenberg, 2003).
Partial reversion was observed in the breast cancer cell lines grown in tissue culture as a disorganized cell mass. Treatment of the 3D culture with an antibody to β1 integrin results in the formation of acini by polarized epithelium and to secretion of milk proteins in response to hormonal stimulation (Schmeichel and Bissell, 2003). Cell proliferation in the acini was significantly inhibited as compared to that in the original culture.
Loss of adhesion of epithelial cells to plastic or basement membrane, realized by integrins, leads to downregulation of epithelial differentiation (see Danen and Sonnenberg, 2003).
Identification of transcription factors which mediate re-differentiation through individual integrin transfection is a key problem in understanding of concrete function and possible reversion via integrins.
4. Matrix metalloproteinases (MMP)
MMPs are tissue enzymes with proteolytic activity, which act in tissue remodeling in ontogenesis and destruction of microenvironment during tumor progression. ECM, including basal membranes, is a primary target of MMPs in tumor progression (Stamenkovic, 2003). ECM components are destructed by MMPs localized on the tumor cell membranes or secreted in the extracellular fluid (Hernandez-Barrahter et al., 2002). Overproduction of MMPs is a very important factor in invasion and metastasis, since they destroy the natural barriers for dissemination of tumor cells into foreign territories, as well as amorphous ECM (Stetler-Stevenson and Yu, 2001).
Destruction of amorphous ECM and basement membrane probably can play another and specific role: MMPs destroy factors necessary for maintaining epithelial differentiation. Moreover, MMPs undoubtedly play an important role in epithelial-mesenchymal trans-differentiation (Section III.E).
Taken together, the data on alterations of cell adhesion in tumor progression show that it is realized through:
- destruction of gap junctional communication and, hence, loss of metabolic cooperation of target tissue;
- disruption (or weakening) of cell-cell contacts;
- disruption or weakening of cell-ECM interactions with loss of epithelial cell polarity and specific functions;
- significant changes in cell-stroma interrelations.
D. Tissue "architecture" in progression: key to tumor markers origin
Tumor markers constitute a special field of oncology, both fundamental and clinical. They include onco-fetal antigens, normal products of the embryo, reappeared in some tumors, such as AFP, or are the result of significant derangement of excretion pathways, as is in the cases of CEA and PSA (see Abelev and Sell, 1999).
Causes of the appearance of a certain marker or significant increase in its blood serum level may be different, but the common event is the change in "tissue architecture" typical for different epithelial tumors.
Earlier it was shown that the formation and integrity of liver plate is necessary for suppression of AFP synthesis in hepatocytes. Liver plate derangement when the hepatocytes are "liberated" from the plate is a necessary condition for AFP gene expression. The presence of 3D ECM and interaction of liver cells with ECM lead to reestablishment of AFP gene suppression (see Abelev and Eraiser, 1999).
There must be initial distinct stages in liver tumor progression. At the initial stage, the tumor tissue retains the morphology of adult liver, with polygonal hepatocytes, with characteristic surface domain distribution and retaining cell-cell interactions, overproduction of intercellular ECM and with obligatory suppression of AFP synthesis. Chain of further stages leads step by step to the loss of epithelial characteristics. Destruction of regular liver plates and cell-ECM interactions, as well as "architectural" derangements in general are obligatory events at the advanced stages of progression. Thus, re-expression of AFP looks like the most expected event. Of course, additional experimental evidence is required to accept this quite logical concept. Anyway, AFP is a widely used serum marker of hepatocellular and germ cell carcinomas (see Abelev and Elgort, 1982).
Another system, where "architectural changes" look most plausible, is CEA increase in the blood of intestinal carcinoma patients (see Hammerström, 1999).
CEA is a component of normal intestinal glycocalyx and is localized strictly on the apical part of intestinal epithelium in the borderbrush. It is normally excreted into the intestinal lumen.
When the epithelial layer of intestine is destroyed, CEA loses its strictly apical localization and can be seen throughout the cytoplasm. It can diffuse into circulation through basolateral surfaces of depolarized epitheliocytes, and that is, most probably, why the blood level of CEA is raised in colorectal cancer patients.
The third marker is PSA. Its increased blood level is a very probable indication of prostate cancer. And again, the most plausible reason is disruption of the normal way of its secretion (Stenman et al., 1999). PSA is kallikrein protease normally secreted to the seminal fluid. In malignant prostatic tissue, the content of dead cells may get into the blood and lead to increased PSA level.
Thus, these three most popular markers appear in blood as a consequence of tumor progression, rather than of expression due to transformation.
E. Epithelial-mesenchymal transition (EMT) as the late step
in progression of epithelial tumors
The most impressive phenomenon in tumor progression is the phenotypic change of malignant epithelium, when cells lose their epithelial polarity, specific epithelial functions, their need in basement membrane, become motile, and begin to express vimentin, typical for motile mesenchymal cells, instead of cytokeratins, typical for epithelium (Thierry, 2002; Gotzman et al., 2004). This transition looks like genetically determined trans-differentiation, but it is rather a morphogenetic event based on changes in the relations of tumor cells with microenvironment.
EMT is relevant to and uses mechanisms similar to those involved in epithelial remodeling in early ontogenesis, which suggests so-called epithelial plasticity. However, EMT most probably leads to invasion and metastases, which are usually associated with the late stages of tumor progression.
It is likely that EMT is associated with the considerable changes in expression of tissue-specific transcription factors and, as a consequence leads to significant or complete epithelial dedifferentiation (Lazarevich et al., 2004). Is it reversible? Is it determined on "one-gene-expression basis" or does it have more complicated nature?
EMT in ras transformed cell lines can be induced by transforming growth factor (TGF) β, which clearly shows that it is not obligatory caused by genetic changes (Zavadil et al., 2001; Gotzman et al., 2002; Stahl and Felsen, 2003; Fischer et al., 2005). Implication of large polyclonal communities of cells in EMT is also in line with physiological (morphogenetic) nature of this phenomenon, rather than gene mutations.
F. Tissue-specific transcription factors in carcinoma progression
1. Dysfunction of HNFs in tumor progression
While the general steps of cellular transformation have been postulated (Hanahan and Weinberg, 2000), each tumor type retains the distinctive features that arise from characteristics of its tissue of origin. The loss of differentiation observed in the majority of epithelial tumors can be due to disbalanced tissue-specific transcriptional network. In this section we will consider the alterations in HNF signaling during epithelial carcinogenesis and/or tumor progression.
Since HNFs are critical for the specification and differentiation of various epithelial cell lineages, it is reasonable to suppose that the dysfunction of these factors is an important determinant of transformation and/or progression of epithelial tumors. Studies of the last decade demonstrated the correlation of alterations in HNF expression and function with epithelial carcinogenesis (summarized in Table 2). Remarkably, the factors playing the most important role in the differentiation of particular tissues are the preferential targets for inactivation in the corresponding tumor types.
| HNFs dysfunction in epithelial tumors | ||||
| Transcription factor | Tumor type | Alteration described | Reversion | Reference |
| HNF1α | Hepatocellular adenoma | Mutation | ND | Bluteau et al., 2002 |
| HNF1α /HNF1β | HCC | Decrease of HNF1α/ HNF1β ratio | ND | Ninomiya et al., 1996 |
| HNF1α | Dedifferentiated HCC | Reduced expression | ND | Wang et al., 1998 |
| HNF1α | Colorectal cancer | Mutation | ND | Laurent-Puig et al., 2003 |
| HNF1α | Renal cell carcinoma | Reduced expression; diminished binding activity | ND | Sel et al., 1996; Anastasiadis et al., 1999 |
| HNF1α and β | Renal cell carcinoma | Germline mutations | ND | Rebouissou et al., 2005 |
| HNF1β | Clear cell carcinoma of the ovary | Upregulation | Apoptotic cell death | Tsuchiya et al., 2003 |
| HNF3β | Lung cancer | Downregulation, mutations, promoter methylation | Growth arrest | Halmos et al., 2004 |
| C/EBPα | HCC | Reduced expression | Growth inhibition, suppression of tumorigenicity | Flodby et al., 1995; Watkins et al., 1996; Xu et al., 2001 |
| C/EBPα | Breast cancer | Reduced expression, cytoplasmic localisation | Growth inhibition, differentiation | Gery et al., 2005 |
| C/EBPα | Lung cancer | Reduced expression | Proliferation arrest, apoptosis, morphological changes | Halmos et al., 2002 |
| C/EBPα | Skin squamous cell carcinoma | Reduced expression | Growth arrest | Shim et al., 2005 |
| C/EBPβ-LIP | Breast cancer | Upregulation | ND | Zahnov et al., 1997 |
| C/EBPβ-2 LAP | Breast cancer | Upregulation | ND | Bundy and Sealy, 2003 |
| C/EBPβ | Ovarian epithelial tumor | Upregulation | ND | Sundfeldt et al., 1999 |
| C/EBPβ | Colorectal cancer | Upregulation | ND | Rask et al., 2000 |
| C/EBPβ | Renal cell carcinoma | Increased activity | ND | Oya et al., 2003 |
| C/EBPβ | Wilms tumor | Upregulation | Apoptosis | Li et al., 2005 |
| HNF4α | Hepatoid adenocarcinoma of stomach | Upregulation | ND | Yano et al., 2003 |
| HNF4α | HCC | Reduced expression | Growth arrest, restoration of epithelial morphology, gene re-expression | Spath and Weiss, 1998; Lazarevich et al., 2004 |
| HNF4α | Renal cell carcinoma | Diminished binding activity | ND | Sel et al., 1996 |
This point can be illustrated by the investigations of HCC progression mechanisms performed in our laboratory in the last few years. HCC is the prevalent liver tumor and one of the world's most common cancers. The major risk factors for the development of HCC are chronic infection with hepatitis B or C viruses and prolonged exposure to hepatocarcinogenes (Bosch et al., 1999). HCC development is a continuous multi-step process that results from the accumulation of genetic alterations leading to the acquisition of more aggressive tumor phenotype. The analysis of genetic abnormalities and gene expression alterations in HCC revealed some candidate genes and signaling pathways possibly implicated in hepatocarcinogenesis (Thorgeirsson and Grisham, 2002). These genes encode growth factors (TGF-α and -β, hepatocyte growth factor (HGF) and their receptors), tumor suppressors (Rb and p53), components of the Wnt/catenin pathway, and other cell communication and adhesion molecules (Buendia, 2000). However, the molecular basis of hepatic tumor progression and its relationship to normal cell differentiation remain obscure.
To elucidate the role of liver-enriched transcription factors in HCC progression, we have developed an experimental model in which a chemically induced slow-growing transplantable mouse HCC (sgHCC) rapidly gave rise in vivo to a highly invasive dedifferentiated fast-growing variant (fgHCC) (Lazarevich et al., 2004).
sgHCC is a highly differentiated tumor with a pattern of gene expression that closely resembles normal hepatocytes. The progression of sgHCC was accompanied by loss of epithelial morphology, activation of telomerase, extinction of liver-specific gene expression, invasion, and ultimately metastasis (Varga et al., 2001; Lazarevich et al., 2004).
The loss of epithelial morphology in fgHCC comprises a complete loss of cell polarity and a decrease in cell-cell and cell-matrix adhesion. sgHCC has a typical basal membrane which consists of type IV collagen, laminin, entactin, and fibronectin; each cell interacts with the basal membrane. In contrast, in fgHCC, all ECM components are synthesized but not associated with the membranes, thus, cell-matrix interactions are completely disarranged. The tight junction marker protein ZO-1 is present on the membrane of sgHCC, while fgHCC cells exhibit no membrane staining of ZO-1. This fact together with the redistribution of domain-specific markers indicates the loss of epithelial polarity in fgHCC (Engelhardt et al., 2000; Kudryavtseva et al., 2001).
The morphological changes observed during a one-step progression meet the criteria of the EMT (Thiery, 2002). It is accompanied by alterations in the expression of several genes coding for the cell-cell and cell-ECM communication components (Lazarevich et al., 2004). Noteworthily, sgHCC, characterized by extremely strong cell-cell and cell-matrix contacts, overexpresses E-cadherin and β-catenin mRNA as compared to normal liver. fgHCC shows significantly decreased steady state mRNA levels of the major liver gap junctional protein Cx32 and E-cadherin, and an overexpressed α3 integrin subunit, which is expressed in immature or transformed hepatocytes and in biliary cells and mediates ECM-induced differentiation (Lora et al., 1998). Downregulation of E-cadherin transcription is accompanied by a significant increase of mRNA steady state level of Snail, a zinc finger transcription factor involved in the epithelial-to-mesenchymal transition through repression of the E-cadherin promoter (reviewed in Peinado et al., 2004). In the mouse HCC model, EMT was also mediated by increased levels of the mesenchymal cell marker vimentin and splice form of fibronectin containing extra domain A (EDA). EDA form of fibronectin promotes the increase of cell spreading and migration (Manabe et al., 1997).
In addition to the loss of epithelial morphology, fgHCC cells demonstrate reduced expression of most markers of mature hepatocytes (albumin, α1-antitrypsin, transthyretin, phenylalanine hydroxylase, apolipoproteins, L-type pyruvate kinase, glucokinase, etc.) as well as the expression of AFP normally restricted to fetal hepatoblasts and induced in sgHCC as compared to normal liver. Thus, according to the expression pattern, the one-step progression caused severe dedifferentiation of fgHCC as compared to sgHCC. These alterations were coupled with the reduced expression of the entire block of liver transcription factors that are essential for hepatocyte differentiation, namely, HNF1α, HNF1β, HNF3γ, C/EBPα, HNF4α (both α1 and α7 isoforms), and HNF6 (Lazarevich et al., 2004). The retained expression of several liver-specific genes in fgHCC confirms the hepatic origin of these cells (Lazarevich et al., 2004).
Multiple phenotypic changes defining the development of a highly malignant phenotype occur rapidly implying that HCC progression in this model is a consequence of a limited set of molecular events. We suppose that HCC progression can result from dysfunction of liver-specific transcription factors which control hepatic gene expression and differentiation. Extensive investigations on animal models indicate that the key liver-specific transcription factors providing for mature hepatic phenotype are HNF4α1, HNF1α, and C/EBPα (Section II.B.1).
The relevance of these factors for the maintenance of hepatic differentiation is confirmed for hepatoma cell cultures. The expression of HNF4α1 as well as of its direct transcriptional target HNF1α is generally restricted to differentiated hepatomas, while HNF1β and HNF4α7 are mainly expressed in dedifferentiated cells (Cereghini et al., 1988; Spath and Weiss, 1998; Nakhei et al., 1998; Torres-Padilla, 2001). In concordance with these data, a significant decrease of HNF1α expression was revealed in poorly differentiated human HCCs relative to well-differentiated tumors (Wang et al., 1998).
Similarly, the expression of C/EBPα, associated with the maintenance of a quiescent differentiated state of hepatocytes, is very low or undetectable in liver nodules and HCC tumor samples (Flodby et al., 1995). Forced expression of C/EBPα in human hepatoma cell lines results in impaired proliferation and suppressed tumorigenicity (Watkins et al., 1996).
The expression of exogenous HNF4α1 in a dedifferentiated hepatoma cell culture H5 induces the expression of several hepatic genes and re-establishment of epithelial cell morphology. This transition is associated with re-expression of cytokeratins and E-cadherin production. These findings indicate that HNF4α may be a key regulator of hepatic differentiation which integrates tissue-specific gene expression and epithelial morphogenesis (Spath and Weiss, 1998).
The studies on the one-step model of HCC progression in mice confirmed the essential role of HNF4α dysfunction in hepatocarcinogenesis (Lazarevich et al., 2004). Forced expression of HNF4α1 in cultured fgHCC cells partially restored epithelial morphology. fgHCC cells expressing HNF4α1 formed epithelial sheets with tight junctions marked by ZO-1 plasma membrane staining. In addition, fibrils of ECM components were located along cell-cell interfaces indicating the formation of cell-matrix contacts. At the same time, E-cadherin was not revealed on the cell membranes of epithelial islands suggesting that epithelial conversion in this case is an E-cadherin independent process.
HNF4α1 re-expression in fgHCC cell culture also restored the expression of several functional hepatic proteins (apolipoproteins, α1-antitrypsin, Cx32, etc.) and transcription factors (HNF1α, HNF6, HNF4α7, and HNF3γ), reduced the proliferation rate in vitro, and dramatically inhibited tumor formation and metastasis in congenic recipient mice. The multiple effects of HNF4α1 on tumor cell characteristics can be mediated by the activation of other HNF4α-regulated liver-enriched transcription factors. Nevertheless, the re-expression of individual transcription factors that were HNF4α-inducible did not result in obvious changes in fgHCC cell proliferation and morphology (Lazarevich, unpublished data).
Thus, the profound changes in the differentiation state and cell proliferation in this model of tumor progression are at least partially defined by HNF4α1 suppression. The restoration of HNF4α1 function can promote the reversion of invasive HCC toward a less aggressive phenotype by inducing hepatic re-differentiation.
The central role of HNF4α1 in the control of morphological, proliferative, and functional properties that define hepatic differentiation was confirmed in other models. Studies of gene expression profiles on the collection of chemically induced mouse HCCs of independent origin revealed a strict correlation between HNF4α1 transcription and tumor differentiation status. Downregulation of HNF4α1 transcription in the fast-growing poorly differentiated tumors coincided with the extinction of transcription factors that previously proved to be HNF4-inducible (HNF1α, HNF4α7, and HNF3γ). We also detected downregulation of HNF4α1 expression in the majority of human HCCs not associated with hepatitis infection (Fleishman and Lazarevich, in preparation). Moreover, a high incidence of dysfunction of HNF4α1 and its transcriptional target HNF1α was revealed in human renal cell carcinomas (Sel et al., 1996; Anastasiadis et al., 1999).
Thus, these factors can be considered as potential tumor suppressor genes at least in the kidney and liver, where HNF4α1/HNF1α regulatory pathway clearly defines the lineage-specific differentiation. These data support the hypothesis that dysfunction of tissue-specific transcription factors essential for definite tissue specification can be a critical step in the dedifferentiation and progression of the corresponding epithelial tumors.
This point can be illustrated by dissecting the role of C/EBP factors in epithelial carcinogenesis. Recently, reduced expression of C/EBPα was described in breast (Gery et al., 2005), lung (Halmos et al., 2002), and skin (Shim et al., 2005) tumors. In lung cancers, the decrease of C/EBPα expression correlates with the histological subtype and tumor grade (Halmos et al., 2002). Restoration of C/EBPα expression in non-small cell lung cancer cell lines induces apoptosis, proliferation arrest, and morphological changes toward a more differentiated phenotype.
mRNA and protein levels of C/EBPα are downregulated in mouse skin squamous cell carcinomas (SCC) and cell lines with activated Ras (Shim et al., 2005). Transduction of keratinocytes with oncogenic ras diminishes the expression and DNA binding of C/EBPα indicating the involvement of activated Ras in negative regulation of C/EBPα transcription. Forced expression of C/EBPα in SCC cell lines inhibits the proliferation of SCC cells containing oncogenic Ras indicating that the antiproliferative activity of C/EBPα is dominant over the Ras effects.
In humans, the normal mammary tissue expresses only C/EBPβ activatory 1 isoform (Bundy and Sealy, 2003). The overexpression of the dominant negative isoform of C/EBPβ induces proliferation and hyperplasia of the mammary gland (Zahnow et al., 2001). In addition, the overexpression of C/EBPβ activatory isoform 2 in mammary epithelial cell line induces EMT and development of invasive phenotype characterized by the loss of E-cadherin junctional localization, acquisition of a spindle-shape morphology, actin stress fibers, and the appearance of the mesenchymal intermediate filament vimentin (Bundy and Sealy, 2003). C/EBPβ regulates the expression of cyclooxygenase 2, the key enzyme of prostaglandin biosynthesis that induces mammary tumorigenesis in transgenic mice and is frequently overexpressed in invasive breast cancers. There is increasing evidence that C/EBPβ acts as downstream effector of Ras signaling. C/EBPβ also plays an important role in Ras-mediated skin tumorigenesis. C/EBPβ -/- mice are refractory to chemically induced skin carcinogenesis (Zhu et al., 2002). All these findings support the idea that dysfunction of tissue-specific transcriptional regulators is an important event in the development of malignant phenotype of different epithelial cell types.
Certainly it is unlikely that the function of the HNF transcriptional network is implemented autonomously from the ubiquitous pathways controlling the growth, morphological, and housekeeping properties of cells. While a significant progress in understanding the impact of HNFs in epithelia differentiation and function was achieved in the last decade, the mutual influence of this tissue-specific transcriptional network and the components of the main signal transduction cascades are still underestimated. In this context, we discuss the presumable mechanisms of HNF4α influence on hepatic differentiation.
What mechanisms define the HNF4-dependent control of hepatic function, epithelial morphology, and proliferation? This question might be addressed by identification of new transcriptional targets of tissue-specific regulatory factors. HNF4α effects on gene regulation can be realized both directly and by regulation of transcription of other HNFs. However, the inactivation of HNFs in mice (Section II.B.1) and their re-expression in dedifferentiated tumors have less dramatic consequences suggesting that the most critical regulation of cell properties is realized by HNF4α per se.
Obviously the major part of experimentally proved HNF4α targets are functional genes encoding blood coagulation factors, enzymes of lipid (Hayhurst et al., 2001), amino acid, glucose (Parviz et al., 2003), and urea (Inoue et al., 2002) metabolism. Importantly, HNF4α regulates the expression of cytochromes P450 and some other enzymes and transporters involved in drug metabolism (reviewed in Tirona and Kim, 2005). Thus, the loss of HNF4α during HCC progression can have an additional significance – HNF4-negative tumors can have altered responsiveness to therapy. In this case, re-expression of HNF4α can be considered as a possible approach to sensitization of HCCs to therapy.
The morphogenic properties of HNF4α are likely realized by transcriptional regulation of several cell adhesion and junction proteins like E-cadherin (Spath and Weiss, 1998; Parviz et al., 2003), Bgp1, gap junctional proteins Cx32 (presumably, through HNF1α regulation) (Piechocki et al., 2000) and Cx26 (Parviz et al., 2003), and tight-junction components occludin and claudins 6 and 7 (Chiba et al., 2003). There is also an evidence for the existence of HNF4-dependent posttranslational mechanisms of ZO-1 dysfunction (Parviz et al., 2003).
HNF4α putative transcriptional targets that can mediate the antiproliferative effects of this factor include p21 (Chiba et al., 2005) and Fas ligand (Osanai et al., 2002). HNF4α is also essential for the hypoxic induction of the erythropoietin gene in the kidney and liver, thus it is likely involved in the cellular oxidative stress response in normal and tumor tissues (Galson et al., 1995).
Additional candidate HNF4α target genes were recently identified using microarray hybridization (Naiki et al., 2002). Chromatin immunoprecipitation analyses combined with promoter microarrays with Hu13K human DNA chip identified about 1500 putative HNF4α target genes in human liver (Odom et al., 2004). The experimental verification of this data will help to disclose the new mechanisms defining the key role of HNF4α in hepatic differentiation.
Studies on the one-step HCC progression model allowed us to demonstrate that the regulatory region of HNF4α1 is inactive in dedifferentiated fgHCC, indicating HNF4α1 repression at the transcriptional level as oppose to mutation, DNA rearrangement, or other damage of the HNF4α1 coding or regulatory regions. This result provides a strong evidence for the existence of HNF4α upstream mechanisms responsible for tumor progression.
2. HNFs mediate microenvironmental effects on hepatic differentiation
It has been clearly demonstrated that the maintenance of epithelial morphology is an important determinant influencing hepatic differentiation. Isolation and cultivation of hepatocytes associated with disruption of normal interaction between cells and ECM leads to drastic changes in the gene expression program. Freshly isolated adult liver hepatocytes, cultivated on dried collagen substrate in the presence of growth factors, actively proliferate, express cytoskeletal components (actin, tubulin, cytokeratins, and vinculin), but exhibit low levels of liver-specific gene expression. Cultivation of hepatocytes on reconstituted ECM gel from Engelbreth-Holm-Swarm mouse sarcoma (EHS gel), primarily composed of laminin, type IV collagen, heparin sulfate proteoglycan matrix, and several growth factors, diminishes DNA synthesis, decreases the transcription of cytoskeletal components, and prevents the loss of liver-specific (albumin, transferrin, and α1-antitrypsin) gene expression (Ben-Ze'ev et al., 1988). What molecular mechanisms translate the extracellular signals into alterations of liver-specific gene expression?
Cultivation of hepatocytes on collagen I decreases HNF4α and C/EBPα expression, while growth on EHS gel maintains the transcriptional level of HNF4α similar to the in vivo level (Oda et al., 1995; Runge et al., 1997). Moreover, dedifferentiated hepatocytes grown on dried collagen I matrix can redifferentiate after the addition of EHS gel. This transition is accompanied by induction of C/EBPα DNA-binding activity (Runge et al., 1997) and HNF4α transcription (Runge et al., 1999). These data suggest that the key factors of hepatic maturation, HNF4α and C/EBPα, can mediate ECM-dependent regulation of liver-specific gene expression. However, the absence of C/EBPα does not prevent ECM-dependent upregulation of albumin gene in primary hepatocytes isolated from C/EBPα -/- mice (Soriano et al., 1998). It is likely that C/EBPα alone is not responsible for this effect.
HNF3 factors are essential for early liver development and activation of liver-specific genes (Lemaigre and Zaret, 2004). HNF3α expression and HNF3 DNA binding activity are upregulated by ECM in the hepatic-derived cell lines H2.35 and HepG2 (DiPersio et al., 1991). In these cell lines, the activation of HNF3 is critical for upregulation of albumin transcription induced by ECM assuming that HNF3 factors can mediate ECM-dependent differentiation. However, the dedifferentiation of primary hepatocytes in culture leads to the upregulation of HNF3α (Oda et al., 1995; Runge et al., 1998), while EHS gel-induced redifferentiation is accompanied by the reduction of HNF3 DNA binding activity (Runge et al., 1998). Thus, it is likely that HNF3 cannot provide ECM-responsive activation of the differentiation program in normal adult hepatocytes. These data are consistent with the observation that the activity of HNF3 proteins has the most profound impact on hepatic differentiation at the early stages of liver development (Sund et al., 2000).
The results supporting the key role of HNFs in ECM-mediated differentiation were obtained on the model of "small hepatocytes" differentiation in culture (Sugimoto et al, 2002). Small hepatocytes are proliferating cells with hepatic characteristics isolated from adult liver. Cultivation in EHS gel induces the changes of the cell shape typical for mature hepatocytes, formation of bile canaliculi-like structures, and production of liver-specific proteins, which correlate with the activation of HNF4α, HNF6, C/EBPα, and C/EBPβ transcription. Addition of individual growth factors, ECM components of the EHS, or collagen gel failed to induce similar alterations in cell morphology and gene expression. It is likely that hepatic differentiation depends on the formation of complex basal membrane-like structures and is modulated by cytokines signaling.
From the first steps of hepatic organogenesis, the paracrine cytokine signaling from mesodermal cells substantially determines the hepatic differentiation into functional hepatocytes (reviewed in Lemaigre and Zaret, 2004). The liver bud formation is induced by fibroblast growth factors (FGFs) and bone morphogenetic proteins (BMPs) produced by the cardiogenic mesoderm and septum transversum, respectively. Further stages of liver development are also mediated by the action of various cytokines produced by stromal cells which control both morphogenesis and proliferation of hepatocytes. Besides, tumor cells, especially those undergoing EMT, gain the ability to secrete growth factors in an autocrine fashion, thus gaining additional independence from the microenvironment (Gotzmann et al., 2002).
The complex interplay between growth factor signaling and ECM effects on the expression of HNFs can be illustrated by studies of in vitro differentiation of fetal mouse hepatocytes. Expression of mature hepatic genes and upregulation of HNF4α in fetal hepatocytes can be induced by EHS and interleukin-6 related hepatic maturation factor oncostatin M (Kamiya et al., 2003). In fetal hepatocytes, this factor directly activates K-Ras and promotes the formation of E-cadherin-based adherence junctions by the Ras-dependent mechanism (Matsui et al., 2002). Hepatic differentiation induced by oncostatin M and EHS can be suppressed by tumor necrosis factor (TNF)-α which inhibits the induction of HNF4α and several liver-specific genes (Kamiya and Gonzalez, 2004).
There is growing evidence that the activity of TGF-β signaling pathway can modulate HNF4α-dependent regulation of hepatic differentiation. HNF4α was shown to physically interact and functionally cooperate with Smad 3 and 4 transcription factors which mediate TGF-β signaling (Chou et al., 2003).
In primary rat hepatocytes, TGF-β was shown to repress HNF4α transcription through activation of WilmTs tumor suppressor WT1, transcription factor referred to as dedifferentiation marker in hepatocytes (Berasain et al., 2003). De Lucas et al. (2004) reported the decrease of HNF4α DNA-binding activity in human hepatoma HepG2 treated with TGF-β and proposed that TGF-β induces HNF4α degradation in the proteasome.
Downregulation of HNF4α expression was also revealed during TGF-β induced EMT in cultured fetal hepatocytes (Sanchez et al., 1999; Valdes et al., 2002) and immortalized hepatocytes transformed with oncogenic Ha-Ras (Gotzmann et al., 2002). These findings suggest that the loss of HNF4α can at least partially mediate the alterations of the functional and morphological properties of hepatocytes during this process.
C/EBP proteins, important mediators of both proliferation and differentiation, are regulated by injury-related cytokines such as TNF-α and interleukin-6 (Diehl, 1998). Suzuki and coworkers (2003) showed that a gradual differentiation of hepatic stem cells to albumin-producing hepatoblasts and finally to mature hepatocytes induced by HGF and oncostatin M is mediated by C/EBPα activation. Lack of C/EBP proteins inhibits hepatic differentiation of stem cells. In this model, several ECM components also induce differentiation by C/EBP expression control, although this effect was less pronounced. Modulation of C/EBP signaling can not only directly influence liver-specific gene expression, but also modify the production of proteins directly involved in the maintenance of epithelial cell morphology. Functional C/EBP sites are identified in the promoters of genes coding ECM proteins, MMPs and protease inhibitors (Doyle et al., 1997; Greenwel et al., 2000; Boffa et al., 2002).
We illustrated the role of ECM in tissue-specific gene regulation on various models of hepatic differentiation; however, it seems suitable for other epithelial tissues. For example, ECM dependent regulation of tissue-specific αs1- and β-casein genes in mammary cells is mediated by alterations in activity of C/EBP proteins, which are critical regulators of mammary gland differentiation (Myers et al., 1998; Jolivet et al., 2005).
Thus, it is likely that tissue-specific transcription factors essential for the specification of definite cell lineages mediate the interaction of epithelial cells with the microenvironment. ECM remodeling, which can regulate signal transduction pathways through interactions with integrins or by releasing growth factors bound to the matrix, together with paracrine signaling from stroma cells can modulate expression or activity of transcription factors. On the other hand, alteration of the transcription factors activity can regulate various genes essential for the maintenance of epithelial morphology. Interrelations between HNF signaling and microenvironment are highly complex and need further investigations, while the importance of tissue-specific transcriptional regulators for the transmission of extracellular signals into alterations of cell transcriptional program is already clear. Consequently, the impairment of tissue-specific transcriptional network seems a very probable cause of epithelial dedifferentiation and carcinoma progression.
IV. Concluding remarks and further perspective
A. Different ways of progression in carcinomas and hemoblastoses
In conclusion, we would like to stress some not trivial features of progression in relation to differentiation in tumors. These features become most clear when epithelial tumors are compared with hemoblastoses. The first and the most crucial difference lies in interrelations between the mechanisms of normal differentiation and development of malignant phenotype in both types of neoplasia. Transformation itself is most probably similar in both systems. It includes constitutive activation of protooncogene or inactivation of tumor suppressor gene and immortalization, i.e. events leading to autonomous and infinite cell proliferation. These processes take place both in hemopoietic and epithelial tissues through the similar mechanisms: protooncogene mutations, activation of protooncogene by its translocation next to functional transcription regulatory elements, or formation of fused genes, driven by the promoters, active in target tissue (see Barr, 1998).
Principal differences appear at the next steps of tumor evolution – in tumor progression – in selection of the most autonomous variants from a genetically unstable population. In epithelial tumors it leads to invasion and metastases, i.e. to ultimate independence on homologous microenvironment permitting the tissue growth in heterologous territories. Dedifferentiation is associated with either process. Thus, partial or complete loss of tissue-specific proteins and antigens is a natural consequence of epithelial tumor evolution.
On the contrary, progression of hemoblastoses tends to maintain the ability of hemopoietic cells of invading normal tissue (diapedesis) and to expand their ability of forming extramedullar islands of clonal growth. Both properties are inherent in the normal hemopoietic tissue. The fatal feature of progression in this system is overproduction of leukemic cells, their accumulation in the bone marrow and blood, extramedullar hemopoesis, and suppression of normal hemopoeisis. However, autonomous leukemic progenitor cells unlike the normal progenitors do not sense the excess of blood cells in leukemia, while normal progenitors are suppressed by this excess.
It should be kept in mind that after differentiation of hemopoietic cells, which takes place in bone marrow microenvironment, committed or mature cells no more require specific microenvironment for maintaining their differentiation state. This is the principal difference between hemopoietic and epithelial tumors, especially in their progression. Progression of hemoblastoses is not only compatible with the normal properties of hemopoietic cells, but is based on their normal physiological functions, thus keeping differentiation antigens unchanged. Rare cases of "promiscuous" hemopoietic line markers expression are due, most probably, to the existence of corresponding transitional stages in development of certain blood cells (Greaves et al. 1986).
B. Future investigations
We are trying to formulate several questions, which remain unresolved on the way of development of a unifying concept, binding microenvironment to epitheliocyte differentiation, and its dedifferentiation in the process of tumor progression due to disruption of microenvironment.
The first problem includes clarification of the microenvironment function in induction versus maintenance of epithelial differentiation. The known elements of this process include obligatory participation of heterologous tissue, most typically mesenchymal tissue, in the specification of epithelial organs, such as liver, pancreas, kidney, mammary gland, skin epidermis, etc. This problem raises the underlying one as to: how tissue interactions induce (and maintain?) a network of tissue-specific transcription factors determining epithelial tissue differentiation. It is obviously a key point of the whole concept, including disruption of these mechanisms in tumor progression. The investigations of Kenneth Zaret and his colleagues as well as George MichalopoulosT team on the regulation of tissue-specific HNFs by ECM gel opened a way in this direction.
The most enigmatic but clearly formulated problem is why and how 3D ECM induces a specific set of transcription factors operating in tissue-specific differentiation? Undoubtedly, this is a key problem relating ECM structure and epitheliocytes differentiation. Its solution is in the air, but the way to solve it is not yet found. Only few publications to our knowledge showed that the nuclear shape and organization of nuclear matrix associated with gene expression depend on ECM structure (Lelievre et al., 1998; Maniotis et al., 1997).
Another very important problem is identification of tissue-specific transcription factors essential for epithelial differentiation. Does epitheliocytes interaction with ECM lead to independent mosaic-like induction of individual transcription factors, or implements through a single crucial step, which determines the further chain of events resulting in the formation of a specific transcriptional network? Evidence of the critical role of HNF4α in hepatocyte differentiation favors the latter possibility (Lazarevich et al., 2004).
How should EMT be regarded: as a regular change in epitheliocytes development, including remodeling of epitheliocytes and natural evolution of tissue structure, or as a result of pathological development of epithelial-mesenchymal interrelations?
And finally, to what extent can tumor progression be reversed? Is the normalization of malignant phenotype limited to some intermediate stages, or can the whole process be turned back? In this respect the identification of definitive events of tumor reversion is extremely important (Mintz and Illmensee, 1975; Kenny and Bissell, 2003).
ACKNOWLEDGMENTS
We would like to acknowledge Prof. Sergey Vassetzky and Dr. Nikita Vassetzky for editing the manuscript. We thank Mrs. Olga Salnikova, Drs. Tatyana Eraiser and Maria Goncharskaya for the help in manuscript preparation.
This work was supported by the program "Leading Russian Scientific Schools", and Russian Foundation for Basic Research, project No 04-04-49189.
References
- Abelev, G. I. (1965). Antigenic structure of chemically induced hepatomas. Prog. Exp. Tumor Res. 7, 104-157.
- Abelev, G. I. (1971). Alpha-fetoprotein in ontogenesis and its association with malignant tumors. Adv. Cancer Res. 14, 295-358.
- Abelev, G. I. (1978). Experimental study of alpha-fetoprotein re-expression in liver regeneration and hepatocellular carcinomas. In "Cell Differentiation and Neoplasia" (G. Saunders, Ed.), pp. 257-269. Raven Press, New York.
- Abelev, G. I. (2000). Differentiation mechanisms and malignancy. Biochemistry (Mosk.) 65, 107-116.
- Abelev, G. I., and Elgort, D. A. (1982). Alpha-Fetoprotein. In: "Cancer Medicine" (J. Holland and E. Frei, Eds.), pp. 1089-1099. Lee and Febiger, New York.
- Abelev, G. I., and Eraizer, T. L. (1999). Cellular aspects of alpha-fetoprotein reexpression in tumors. Semin. Cancer Biol. 9, 95-108.
- Abelev, G. I., and Sell, S. (1999). Introduction. Semin. Cancer Biol. (Tumor Markers) 9, 61-66.
- Aggeler, J., Park, C. S., and Bissell, M. J. (1988). Regulation of milk protein and basement membrane gene expression: the influence of the extracellular matrix. J. Dairy Sci. 71, 2830-2842.
- Akiyama, T. E., Ward, J. M., and Gonzalez, F. J. (2000). Regulation of the liver fatty acid-binding protein gene by hepatocyte nuclear factor 1alpha (HNF1alpha). Alterations in fatty acid homeostasis in HNF1alpha-deficient mice. J. Biol. Chem. 275, 27117-27122.
- Anastasiadis, A. G., Lemm, I., Radzewitz, A., Lingott, A., Ebert, T., Ackermann, R., Ryffel, G. U., and Schulz, W. A. (1999). Loss of function of the tissue specific transcription factor HNF1 alpha in renal cell carcinoma and clinical prognosis. AntiCancer Res. 19, 2105-2110.
- Andriani, F., Garfield, J., Fusenig, N. E., and Garlick, J. A. (2003). Basement membrane proteins promote progression of intraepithelial neoplasia in 3-dimensional models of human stratified epithelium. Int. J. Cancer 108, 348-357.
- Ang, S. L., and Rossant, J. (1994). HNF-3 beta is essential for node and notochord formation in mouse development. Cell 78, 561-574. 2735-2766.
- Ang, S. L., Wierda, A., Wong, D., Stevens, K. A., Cascio, S., Rossant, J., and Zaret, K. S. (1993). The formation and maintenance of the definitive endoderm lineage in the mouse: involvement of HNF3/forkhead proteins. Development 119, 1301-1315.
- Arias, A. M. (2001). Epithelial mesenchimal interactions in cancer and development. Cell 105, 425-431.
- Barbacci, E., Reber, M., Ott, M.O., Breillat, C., Huetz, F., and Cereghini, S. (1999). Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development 126, 4795-4805.
- Barr, F. G. (1998). Translocations, cancer and puzzle of specificity. Nature 19, 121-124.
- Behrens, J., Vakaet, L., Friis, R., Winterhager, E., Van Roy, F., Mareel, M. M., and Birchmeier, W. (1993). Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-src gene. J. Cell Biol. 120, 757-766.
- Ben-Ze'ev, A., Robinson, G. S., Bucher, N. L., and Farmer, S. R. (1988). Cell-cell and cell-matrix interactions differentially regulate the expression of hepatic and cytoskeletal genes in primary cultures of rat hepatocytes. Proc. Natl. Acad. Sci. USA 85, 2161-2165.
- Berasain, C., Herrero, J. I., Garcia-Trevijano, E. R., Avila, M. A., Esteban, J. I., Mato, J. M., and Prieto, J. (2003). Expression of Wilms' tumor suppressor in the liver with cirrhosis: relation to hepatocyte nuclear factor 4 and hepatocellular function. Hepatology 38, 148-157.
- Berenblum, I. (1954). A speculative review; the probable nature of promoting action and its significance in the understanding of the mechanism of carcinogenesis. Cancer Res. 14, 471-477.
- Bissell, M. J., Radisky, D. C., Rizki, A., Weaver, V. M., and Petersen, O. W. (2002). The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70, 537-546.
- Bluteau, O., Jeannot, E., Bioulac-Sage, P., Marques, J. M., Blanc, J. F., Bui, H., Beaudoin, J. C., Franco, D., Balabaud, C., Laurent-Puig, P., and Zucman-Rossi, J. (2002). Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat. Genet. 32, 312-315.
- Boffa, M. B., Hamill, J. D., Bastajian, N., Dillon, R., Nesheim, M. E., and Koschinsky, M. L. (2002). A role for CCAAT/enhancer-binding protein in hepatic expression of thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 277, 25329-25336.
- Bort, R., Martinez-Barbera, J. P., Beddington, R. S., and Zaret, K. S. (2004). Hex homeobox gene-dependent tissue positioning is required for organogenesis of the ventral pancreas. Development 131, 797-806.
- Bosch F. X., Ribes, J., and Borras, J. (1999). Epidemiology of primary liver cancer. Semin. Liver Dis. 19, 271-285.
- Braun, S., Pantel, K., Müller, P., Janni, W., Hepp, F., Kentenich, R. M., Gastroph, S., Wischnik, A., Dimpfl, T., Kindermann, G., Riethmüller, G., and Schlimok, G. (2000). Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N. Engl. J. Med. 342, 525-533.
- Briancon, N., Bailly, A., Clotman, F., Jacquemin, P., Lemaigre, F. P., and Weiss, M. C. (2004). Expression of the alpha7 isoform of hepatocyte nuclear factor (HNF) 4 is activated by HNF6/OC-2 and HNF1 and repressed by HNF4alpha1 in the liver. J. Biol. Chem. 279, 33398-33408.
- Brill, S., Zvibel, I., Halpern, Z., and Oren, R. (2001). The role of fetal and adult hepatocyte extracellular matrix in the regulation of tissue-specific gene expression in fetal and adult hepatocytes. Eur. J. Cell. Biol. 81, 43-50.
- Buendia, M. A. (2000). Genetics of hepatocellular carcinoma. Semin. Cancer Biol. 10, 185-200.
- Bundy, L. M., and Sealy, L. (2003). CCAAT/enhancer binding protein beta (C/EBPbeta)-2 transforms normal mammary epithelial cells and induces epithelial to mesenchymal transition in culture. Oncogene 22, 869-883.
- Cavallaro, U., and Christofori, G. (2004). Cell adhesion and signaling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 4, 118-132.
- Cereghini, S. (1996). Liver-enriched transcription factors and hepatocyte differentiation. FASEB J. 10, 267-282.
- Cereghini, S., Blumenfeld, M., and Yaniv, M. (1988). A liver-specific factor essential for albumin transcription differs between differentiated and dedifferentiated rat hepatoma cells. Genes Dev. 2, 957-974.
- Chandrasekaran, C., and Gordon, J. I. (1993). Cell lineage-specific and differentiation-dependent patterns of CCAAT/enhancer binding protein alpha expression in the gut epithelium of normal and transgenic mice. Proc. Natl. Acad. Sci. USA 90, 8871-8875.
- Chen, W. S., Manova, K., Weinstein, D. C., Duncan, S. A., Plump, A. S., Prezioso, V. R., Bachvarova, R. F., and Darnell, J. E., Jr. (1994). Disruption of the HNF4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 8, 2466-2477.
- Chiba, H., Gotoh, T., Kojima, T., Satohisa, S., Kikuchi, K., Osanai, M., and Sawada, N. (2003). Hepatocyte nuclear factor (HNF)-4alpha triggers formation of functional tight junctions and establishment of polarized epithelial morphology in F9 embryonal carcinoma cells. Exp. Cell. Res. 286, 288-297.
- Chiba, H., Itoh, T., Satohisa, S., Sakai, N., Noguchi, H., Osanai, M., Kojima, T., and Sawada, N. (2005). Activation of p21CIP1/WAF1 gene expression and inhibition of cell proliferation by overexpression of hepatocyte nuclear factor-4alpha. Exp. Cell Res. 302, 11-21.
- Chou, W. C., Prokova, V., Shiraishi, K., Valcourt, U., Moustakas, A., Hadzopoulou-Cladaras, M., Zannis, V. I., and Kardassis, D. (2003). Mechanism of a transcriptional cross talk between transforming growth factor-beta-regulated Smad3 and Smad4 proteins and orphan nuclear receptor hepatocyte nuclear factor-4. Mol. Biol. Cell. 14, 1279-1294.
- Chrenek, M. A., Wong, P., and Weaver, V. M. (2001). Tumor-stromal interactions: integrins and cell adhesions as modulators of mammary cell survival and transformation. Breast Cancer Res. 3, 224-229.
- Christofori, G., and Semb, H. (1999). The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 24, 73-76.
- Cirillo, L. A., Lin, F. R., Cuesta, I., Friedman, D., Jarnik, M., and Zaret, K. S. (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279-289.
- Clark, K. L., Halay, E. D., Lai, E., and Burley, S. K. (1993). Co-crystal structure of the HNF3/fork head DNA-recognition motif resembles histone H5. Nature 364, 412-420.
- Clotman, F., Lannoy, V. J., Reber M., Cereghini, S., Cassiman, D., Jacquemin, P., Roskams, T., Rousseau, G. G., and Lemaigre, F. P. (2002). The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development 129, 1819-1828.
- Clotman, F., Libbrecht, L., Gresh, L., Yaniv, M., Roskams, T., Rousseau, G. G., and Lemaigre, F. P. (2003). Hepatic artery malformations associated with a primary defect in intrahepatic bile duct development. J. Hepatol. 39, 686-692.
- Coffinier, C., Thepot, D., Babinet, C., Yaniv, M., and Barra, J. (1999). Essential role for the homeoprotein vHNF1/HNF1beta in visceral endoderm differentiation. Development 126, 4785-4794.
- Coffinier, C., Gresh, L., Fiette, L., Tronche, F., Schutz, G., Babinet, C., Pontoglio, M., Yaniv, M., and Barra, J. (2002). Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development 129, 1829-1838.
- Costa, R. H., Grayson, D. R., and Darnell, J. E., Jr. (1989). Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and α1-antitrypsin genes. Mol. Cell. Biol. 9, 1415-1425.
- Costa, R. H., Kalinichenko, V. V., and Lim, L. (2001). Transcription factors in mouse lung development and function. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L823-838.
- Cukierman, E., Pankov, R., Stevens, D. R., and Yamada, K. M. (2001). Taking cell-matrix adhesions to the third dimension. Science 294, 1708-1712.
- Cukierman, E., Pankov, R., and Yamada, K. M. (2002). Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 14, 633-639.
- Cumbiner, B. M. (1996). Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84, 345-357.
- Cunha, G. R., Hayward, S. W., Wang, Y. Z., and Ricke, W. A. (2003). Role of the stromal microenvironment in carcinogenesis of the prostate. Int. J. Cancer 107, 1-10.
- Dahl, U., Sjodin, A., and Semb, H. (1996). Cadherins regulate aggregation of pancreatic b-cells in vivo. Development 122, 2895-2902.
- Danen, E. H. J., and Sonnenberg, A. (2003). Integrins in regulation of tissue development and function. J. Pathology 200, 471-480.
- Daniels, E., Letourneau, S., Turbide, C., Kuprina, N., Rudinskaya, T., Yazova, A., Holmes, K. V., Dveksler, G. S., and Beauchemin, N. (1996). Biliary glycoprotein 1 expression during embryogenesis: correlation with events of epithelial differentiation, mesenchymal-epithelial interactions, absorption, and myogenesis. Devel. Dynamics 206, 272-290.
- De Lucas, S., Lopez-Alcorocho, J. M., Bartolome, J., and Carreno, V. (2004). Nitric oxide and TGF-b1 inhibit HNF-4a function in HepG2 cells. Biochem. Biophys. Res. Commun. 321, 688-694.
- De Simone, V., De Magistris, L., Lazzaro, D., Gestner, J., Monaci, P., Nicosia, A., and Cortese, R. (1991). LFB3, a heterodimer forming homeoprotein of the LFB1 family, is expressed in specialized epithelia. EMBO J. 5, 1435-1443.
- De Wever, O., and Mareel, M. (2000). Role of tissue stroma in cancer cell invasion. J. Pathol. 200, 429-447.
- Deininger, M. W. N., Goldman, J. M., and Melo, J. V. (2000). The molecular biology of chronic myeloid leukemia. Blood 96, 3343-3356.
- Descombes, P., and Schibler, U. (1991). A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67, 569-579.
- Diehl, A. M. (1998). Roles of CCAAT/enhancer-binding proteins in regulation of liver regenerative growth. J. Biol. Chem. 273, 30843-30846.
- DiPersio, C. M., Jackson, D. A., and Zaret, K. S. (1991). The extracellular matrix coordinately modulates liver transcription factors and hepatocyte morphology. Mol. Cell. Biol. 11, 4405-4414.
- Dotto, G. P., Parada, L. F., and Weinberg, R. A. (1985). Specific growth response of ras-transformed embryo fibroblasts to tumour promoters. Nature 318, 472-475.
- Doyle, G. A. R., Pierce, R. A., and Parks, W. C. (1997). Transcriptional induction of collagenase-1 in differentiated monocyte-like (U937) cells is regulated by AP-1 and an upstream C/EBP-beta site. J. Biol. Chem. 272, 11840-11849.
- Dufort, D., Schwartz, L., Harpal, K., and Rossant, J. (1998). The transcription factor HNF3b is required in visceral endoderm for normal primitive streak morphogenesis. Development 125, 3015-3025.
- Duncan, S. A., Nagy, A., and Chan, W. (1997). Murine gastrulation requires HNF-4 regulated gene expression in the visceral endoderm: tetraploid rescue of Hnf-4(-/-) embryos. Development 124, 279-287.
- Edlund, H. (2001). Developmental biology of the pancreas. Diabetes 50, S5-9.
- El-Sabban, M.,E., Sfeir, A. J., Daher, M. H., Kalaany, N. Y., Bassam, R. A., and Talhouk, R. S. (2003). ECM-induced gap junctional communication enhances mammary epithelial cell differentiation. J. Cell Science 116, 3531-3541.
- Engelhardt, N. V, Kudryavtseva, E. I., Morozova, O. V., Rudinskaya, T. D., and Turusov, V. S. (2000). Rapid progression of transplantable mouse hepatocarcinoma associated with the loss of cell-surface polarity. Arkh. Patol. (Russ.) 62, 24-29.
- Engelhardt, N. V., Baranov, V. N., Lazareva, M. N., and Goussev, A. I. (1984). Ultrastructural localization of alpha-fetoprotein (AFP) in regenerating mouse liver poisoned with CCl4. Reexpression of AFP in differentiated hepatocytes. Histochemistry 80, 401-407.
- Fischer, A. N., Herrera, B., Mikula, M., Proell, V., Fuchs, E., Gotzmann, J., Schulte-Hermann, R., Beug, H., Mikulits, W. (2005). Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis. Carcinogenesis 26, 931-942.
- Flodby, P., Barlow, C., Kylefjord, H., Ahrlund-Richter, L., and Xanthopoulos, K. G. (1996). Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein alpha. J. Biol. Chem. 271, 24753-24760.
- Galson, D. L., Tsuchiya, T., Tendler, D. S., Huang, L. E., Ren, Y., Ogura, T., and Bunn, H. F. (1995). The orphan receptor hepatic nuclear factor 4 functions as a transcriptional activator for tissue-specific and hypoxia-specific erythropoietin gene expression and is antagonized by EAR3/COUP-TF1. Mol. Cell. Biol. 15, 2135-2144.
- Gao, N., Zhang, J., Rao, M. A., Case, T. C., Mirosevich, J., Wang, Y., Jin, R., Gupta, A., Rennie, P. S., and Matusik, R. J. (2003). The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 17, 1484-1507.
- Gery, S., Tanosaki, S., Bose, S., Bose, N., Vadgama, J., and Koeffler, H. P. (2005). Down-regulation and growth inhibitory role of C/EBPalpha in breast cancer. Clin. Cancer Res. 11, 3184-3190.
- Giancotti, F. G., and Ruoslahti, E. (1999). Integrin signaling. Science 285, 1028-1032.
- Giles, F. J., Keating, A., Goldstone, A. H., Avivi, I., Willman, C. L., and Kantarjian, H. M. (2002). Acute Myeloid leukemia. Hematology 1, 73-110.
- Gleiberman, L., and Abelev, G. (1985). Cell position and cell interactions in expression of fetal phenotype of hepatocyte. Int. Rev. Cytol. 95, 229-266.
- Gleiberman, A. S., Sharovskaya, Yu. Yu., and Chailakhjan, L. M. (1989a) "Contact inhibition" of α-fetoprotein synthesis and junctional communication in adult mouse hepatocyte culture. Exp. Cell Res. 184, 228-234.
- Gleiberman, A. S., Kudrjavtseva, E. I., Sharovskaya, Yu. Yu., and Abelev, G. I. (1989 b). The synthesis of alpha-fetoprotein in hepatocytes is coordinately regulated with cell-cell and cell-matrix interactions. Mol. Biol. Med. 6, 95-107.
- Gotzmann, J., Huber, H., Thallinger, C., Wolschek, M., Jansen, B., Schulte-Hermann, R., Beug, H., and Mikulits, W. (2002) Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-β1 and Ha-Ras: steps towards invasiveness. J. Cell Science 115, 1189-1202.
- Gotzmann, J., Mikula, M., Eger, A., Schulte-Hermann, R., Foisner, R., Beug, H., and Mikulits, W. (2004). Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat. Res. 566, 9-20.
- Greaves, M. F. (1982). Leukemogenesis and differentiation: A commentary. Cancer Surveys 1, 189-204.
- Greaves, M. F., Chan, L. C., Furley, A. J., Watt, S. M., and Molgaard, H. V. (1986). Lineage promiscuity in hemopoietic differentiation and leukemia. Blood 67, 1-11.
- Greenbaum, L. E., Cressman, D. E., Haber, B. A., and Taub, R. (1995). Coexistence of C/EBP alpha, beta, growth-induced proteins and DNA synthesis in hepatocytes during liver regeneration. Implications for maintenance of the differentiated state during liver growth. J. Clin. Invest. 96, 1351-1365.
- Greenbaum, L. E., Li, W., Cressman, D. E., Peng, Y., Ciliberto, G., Poli, V., and Taub, R. (1998). CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J. Clin. Invest. 102, 996-1007.
- Greenwel, P., Tanaka, S., Penkov, D., Zhang, W., Olive, M., Moll, J., Vinson, C., Di Liberto, M., and Ramirez, F. (2000). Tumor necrosis factor alpha inhibits type I collagen synthesis through repressive CCAAT/enhancer-binding proteins. Mol. Cell. Biol. 20, 912-918.
- Gresh, L., Fischer, E., Reimann, A., Tanguy, M., Garbay, S., Shao, X., Hiesberger, T., Fiette, L., Igarashi, P., Yaniv, M., and Pontoglio, M. (2004). A transcriptional network in polycystic kidney disease. EMBO J. 23, 1657-1668.
- Guelstein, V. I., and Khramkova, N. I. (1965). Antigenic structure of mouse hepatomas. VI. Comparison of the antigenic structure of induced hepatomas and their transplants of the first generation. Neoplasma 12, 251-260.
- Guguen-Guillouzo, T., Clement, B., Baffer, G., Beammont, C., Morel-Chany, E., Glaise, D., and Guillouzo, A. (1983). Maintenance and reversibility of active albumin secretion by adult rat hepatocytes co-cultured with another liver epithelial cell type. Exp. Cell Res. 143, 47-54.
- Guillouzo, A., Morel, F., Fardel, O., and Meunier, B. (1993). Use of human hepatocyte cultures for drug metabolism studies. Toxicology 82, 209-219.
- Gupta, R. K., Vatamaniuk, M. Z., Lee, C. S., Flaschen, R. C., Fulmer J. T., Matschinsky, F. M., Duncan, S. A., and Kaestner, K. H. (2005). The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J. Clin. Invest. 115, 1006-1015.
- Habener, J. F., Kemp, D. M., and Thomas, M. K. (2005). Transcriptional regulation in pancreatic development. Endocrinology 146, 1025-1034.
- Hahn, W. C., and Weinberg, R. A. (2002). Modelling the molecular circuitry of cancer. Nat. Rev. Cancer 2, 331-341.
- Halmos, B., Huettner, C. S., Kocher, O., Ferenczi, K., Karp, D. D., and Tenen, D. G. (2002). Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res. 62, 528-534.
- Halmos, B., Basseres, D. S., Monti, S., D'Alo, F., Dayaram, T., Ferenczi, K., Wouters, B. J., Huettner, C. S., Golub, T. R., and Tenen, D. G. (2004). A transcriptional profiling study of CCAAT/enhancer binding protein targets identifies hepatocyte nuclear factor 3 beta as a novel tumor suppressor in lung cancer. Cancer Res. 64, 4137-4147.
- Hammarström, S. (1999). The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 9, 67-82.
- Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57-70.
- Haumaitre, C., Barbacci, E., Jenny, M., Ott, M. O., Gradwohl, G., and Cereghini, S. (2005). Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc. Natl. Acad. Sci. USA 102, 1490-1495.
- Hayhurst, G. P., Lee, Y. H., Lambert, G., Ward, J. M., and Gonzalez F. J. (2001). Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21, 1393-1403.
- Hennighausen, L., and Robinson, G. W. (1998). Think globally, act locally: the making of a mouse mammary gland. Genes Dev. 12, 449-455.
- Hernandez-Barrantes, S., Bernando, M., Toth, M., and Fridman, R. (2002). Regulation of membrane type-matrix metalloproteinases. Semin. Cancer Biol. 12, 131-138.
- Holst, C. R., Nuovo, G. J., Esteller, M., Chew, K., Baylin, S. B., Herman, J. G., and Tlsty, T. D. (2003). Methylation of p16INK4a promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res. 63, 1596-1601.
- Hynes, R. O. (2002). Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673-687.
- Igarashi, P. (2003). Following the expression of a kidney-specific gene from early development to adulthood. Nephron. Exp. Nephrol. 94, e1-6.
- Inoue, Y., Hayhurst, G. P., Inoue, J., Mori, M., and Gonzalez, F. J. (2002). Defective ureagenesis in mice carrying a liver-specific disruption of hepatocyte nuclear factor 4alpha (HNF4alpha). HNF4alpha regulates ornithine transcarbamylase in vivo. J. Biol. Chem. 277, 25257-25265.
- Jacquemin, P., Durviaux, S. M., Jensen, J., Godfraind, C., Gradwohl, G., Guillemot, F., Madsen, O. D., Carmeliet, P., Dewerchin, M., Collen, D., Rousseau, G. G., and Lemaigre F. P (2000). Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol. Cell. Biol. 20, 4445-4454.
- Javaherian, A., Vaccariello, M., Fusenig, N. F., and Garlick, J. A. (1998). Normal keratinocytes suppress early stages of neoplastic progression in stratified epithelium. Cancer Res. 58, 2200-2208.
- Jiang, G., Nepomuceno, L., Yang, Q., and Sladek, F. M. (1997). Serine/threonine phosphorylation of orphan receptor hepatocyte nuclear factor 4. Arch. Biochem. Biophys. 340, 1-9.
- Jochheim, A., Hillemann, T., Kania, G., Scharf, J., Attaran, M., Manns, M. P., Wobus, A. M., and Ott, M. (2004). Quantitative gene expression profiling reveals a fetal hepatic phenotype of murine ES-derived hepatocytes. Int. J. Dev. Biol. 48, 23-29.
- Jolivet, G., Pantano, T., and Houdebine, L. M. (2005). Regulation by the extracellular matrix (ECM) of prolactin-induced alphas1-casein gene expression in rabbit primary mammary cells: Role of STAT5, C/EBP, and chromatin structure. J. Cell. Biochem. 95, 313-327.
- Kaestner, K. H., Hiemisch, H., and Schutz, G. (1998). Targeted disruption of the gene encoding hepatocyte nuclear factor 3gamma results in reduced transcription of hepatocyte-specific genes. Mol. Cell. Biol. 18, 4245-4251.
- Kaestner, K. H., Katz, J., Liu, Y., Drucker, D. J., and Schutz, G. (1999). Inactivation of the winged helix transcription factor HNF3a affects glucose homeostasis and islet glucagons gene expression in vivo. Genes Dev. 13, 495-504.
- Kamiya A., and Gonzalez, F. J. (2004). TNF-alpha regulates mouse fetal hepatic maturation induced by oncostatin M and extracellular matrices. Hepatology 40, 527-536.
- Kamiya, A., Inoue, Y., and Gonzalez, F. J. (2003). Role of the hepatocyte nuclear factor 4alpha in control of the pregnane X receptor during fetal liver development. Hepatology 37, 1375-1384.
- Karen, J., Javaherian, A., Vaccariello, M., Fusenig, N. E., and Garlick, J. A. (1999). 12-O-tetradecanoylphorbol-13-acetate induces clonal expansion of potentially malignant keratinocytes in a tissue model of early neoplastic progression. Cancer Res. 59, 474-481.
- Kenny, P. A., and Bissel, M. J. (2003). Tumor reversion: Correction of malignant behavior by microenvironmental cues. Int. J. Cancer 107, 688-695.
- Khramkova, N. I., and Guelstein, V. I. (1965). Antigenic structure of mouse hepatomas. V. Organospecific liver antigens and embryonic α-globulin in hepatomas of mice induced with orthoaminoazotoluene (AAT). Neoplasma 12, 239-250.
- Kim, S. K., and Hebrok, M. (2001). Intercellular signals regulating pancreas development and function. Genes. Dev. 15, 111-127.
- Krutovskikh, V. (2002). Implication of direct host-tumor intercellular interactions in non-immune host resistance to neoplastic growth. Semin. Cancer Biol. 12, 267-276.
- Ku, N-O., Zhou, X., Toivola, D. M., and Omary, M. B. (1999). The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 227, G1108-G1137.
- Kudryavtseva, E. I, Morozova, O. V, Rudinskaya, T. D, and Engelhardt, N. V. (2001). Disturbances of intercellular contacts and interaction of cells with extracellular matrix in the rapidly growing mouse hepatocarcinoma. Arkh. Patol. (Russ.) 63, 33-37.
- Kudryavtseva, E. I., and Engelhardt, N. V. (2003). Requirement of 3D extracellular network for maintenance of mature hepatocyte morphology and suppression of alpha-fetoprotein synthesis in vitro. Immunol. Lett. 90, 25-31.
- Kuo, C. J., Conley, P. B., Chen, L., Sladek, F. M., Darnell, J. E., Jr., and Crabtree, G. R. (1992). A transcriptional hierarchy involved in mammalian cell-type specification. Nature 355, 457-461.
- Kuperwasser, C., Chavarria, T., Wu, M., Magrane, C., Gray, J. W., Carey, L., Richardson, A., and Weinberg, R. A. (2004). Reconstruction of functionally normal and malignant human breast tissue in mice. Proc. Nat. Acad. Sci. USA 101, 4966-4971.
- Kuprina, N. I., Baranov, V. N., Yazova, A. K., Rudinskaya, T. D., Escribano, M., Cardies, J., Gleiberman, A. S., and Goussev, A. I. (1990). The antigen of bile canaliculi of the mouse hepatocyte: identification and ultr�structural localization. Histochemistry 94, 179-186.
- Kuprina, N. I., Gleiberman, A. S., Beloshapkina-Rudinskaya, T. D., and Abelev, G. I. (1985). Alpha-fetoprotein synthesis in relation to structural peculiarities in postnatal and regenerating mouse liver. Int. J. Cancer 35, 85-92.
- Laconi, S., Pillai, S., Porcu, P. P., Shafritz, D. A., Pani, P., and Laconi, E. (2001a). Massive liver replacement by transplanted hepatocytes in the absence of exogenous growth stimuli in rats treated with retrorsine. Am. J. Pathol. 158, 771-777.
- Laconi, S., Pani, P., Pillai, S., Pasciu, D., Sarma, D. S. R., and Laconi, E. (2001b). A growth-constrained environment drives tumor progression in vivo. Proc. Natl. Acad. Sci. USA 98, 7807-7811.
- Lai, E., Prezioso, V. R., Smith, E., Litvin, O., Costa, R. H., and Darnell, J. E., Jr. (1990). HNF-3A, a hepatocyte-enriched transcription factor of novel structure is regulated transcriptionally. Genes Dev. 4, 1427-1436.
- Landschulz, W. H., Johnson, P. F., and McKnight, S. L. (1988). The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240, 1759-1764.
- Laurent-Puig, P., Plomteux, O., Bluteau, O., Zinzindohoue, F., Jeannot, E., Dahan, K., Kartheuser, A., Chapusot, C., Cugnenc, P. H., and Zucman-Rossi, J. (2003). Frequent mutations of hepatocyte nuclear factor 1 in colorectal cancer with microsatellite instability. Gastroenterology 124, 1311-1314.
- Lazarevich, N. L. (2000). Molecular mechanisms of alpha-fetoprotein gene expression. Biochemstry (Moscow) 65, 117-133.
- Lazarevich, N. L., Cheremnova, O. A., Varga, E. V., Ovchinnikov, D. A., Kudrjavtseva, E. I., Morozova, O. V., Fleishman, D. I., Engelhardt, N. V., and Duncan, S. A. (2004). Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 39, 1038-1047.
- Lazzaro, D., De Simone, V., De Magistris, L., Lehtonen, E., and Cortese, R. (1992). LFB1 and LFB3 homeoproteins are sequentially expressed during kidney development. Development 114, 469-479.
- Lee, C. S., Sund, N. J., Behr, R., Herrera, P. L., and Kaestner, K. H. (2005). Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev. Biol. 278, 484-495.
- Lelievre, S. A., Weaver, V. M., Nickerson, J. A., Larabell, C. A., Bhaumik, A., Petersen, O. W., and Bissell, M. J. (1998). Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus. Proc. Natl. Acad. Sci. USA 95, 14711-14716.
- Lemaigre, F. P., Durviaux, S. M., Truong, O., Lannoy, V. J., Hsuan, J. J., and Rousseau, G. G. (1996). Hepatocyte nuclear factor 6, a transcription factor that contains a novel type of homeodomain and a single cut domain. Proc. Natl. Acad. Sci. USA 93, 9460-9464.
- Lemaigre, F., and Zaret, K. S. (2004). Liver development update: new embryo models, cell lineage control, and morphogenesis. Curr. Opin. Genet. Dev. 14, 582-590.
- Li, J., Ning, G., and Duncan, S. A. (2000). Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev. 14, 464-474.
- Li, W., Kessler, P., Yeger, H., Alami, J., Reeve, A. E., Heathcott, R., Skeen, J., and Williams, B. R. G. (2005). A gene expression signature for relapse of primary Wilms tumors. Cancer Res. 65, 2592-2601.
- Locker, J. (2001). Tissue-specific regulation by transcription factors. In "Transcription factors" (J. Locker, Ed.), pp. 237-262. BIOS Scientific Publishers Ltd, Oxford.
- Lora, J. M., Rowader, K. E., Soares, L., Giancotti, F., and Zaret, K.S. (1998). Alpha3beta1 integrin as a critical mediator of the hepatic differentiation response to the extracellular matrix. Hepatology 28, 1095-1104.
- Manabe, R., Ohe, N., Maeda, T., Fukuda, T., and Sekiguchi, K. (1997). Modulation of cell-adhesive activity of fibronectin by the alternatively spliced EDA segment. J. Cell. Biol. 139, 295-307.
- Maniotis, A. J., Chen, C. S., and Ingber, D. E. (1997). Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA 94, 849-854.
- Matsui, T., Kinoshita, T., Morikawa, Y., Tohya, K., Katsuki, M., Ito, Y., Kamiya, A., and Miyajima, A. (2002). K-Ras mediates cytokine-induced formation of E-cadherin-based adherens junctions during liver development. EMBO J. 21, 1021-1030.
- Matsumura, T., Makino, R., and Mitamura, K. (2001). Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clinical Cancer Res. 7, 594-599.
- McPherson, C. E., Shim, E. Y., Friedman, D. S., and Zaret, K. S. (1993). An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell 75, 387-398.
- Mintz, B., and Illmensee, K. (1975). Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 72, 3585-3589.
- Monaghan, A. P., Kaestner, K. H., Grau, E., and Schutz, G. (1993). Postimplantation expression patterns indicate a role for the mouse forkhead/HNF-3 alpha, beta and gamma genes in determination of the definitive endoderm, chordamesoderm and neuroectoderm. Development 119, 567-578.
- Moorman, A. F., de Boer, P. A., Evans, D., Charles, R., and Lamers, W. H. (1990). Expression patterns of mRNAs for alpha-fetoprotein and albumin in the developing rat: the ontogenesis of hepatocyte heterogeneity. Histochem. J. 22, 653-660.
- Murray, A. W., and Fitzgerald, D. J. (1979). Tumor promoters inhibit metabolic cooperation in cocultures of epidermal and 3T3 cells. Biochem. Biophys. Res. Commun. 91, 395-401.
- Myers, C. A., Schmidhauser, C., Mellentin-Michelotti, J., Fragoso, G., Roskelley, C. D., Casperson, G., Mossi, R., Pujuguet, P., Hager, G., and Bissell, M. J. (1998). Characterization of BCE-1, a transcriptional enhancer regulated by prolactin and extracellular matrix and modulated by the state of histone acetylation. Mol. Cell. Biol. 18, 2184-2195.
- Naiki, T., Nagaki, M., Shidoji, Y., Kojima, H., Imose, M., Kato, T., Ohishi, N., Yagi, K., and Moriwaki, H. (2002). Analysis of gene expression profile induced by hepatocyte nuclear factor 4alpha in hepatoma cells using an oligonucleotide microarray. J. Biol. Chem. 277, 14011-14019.
- Nakhei, H., Lingott, A., Lemm, I., Ryffel, G. U. (1998). An alternative splice variant of the tissue specific transcription factor HNF4alpha predominates in undifferentiated murine cell types. Nucleic Acids Res. 26, 497-504.
- Ninomiya, T., Hayashi, Y., Saijoh, K., Ohta, K., Yoon, S., Nakabayashi, H., Tamaoki, T., Kasuga, M., and Itoh, H. (1996). Expression ratio of hepatocyte nuclear factor-1 to variant hepatocyte nuclear factor-1 in differentiation of hepatocellular carcinoma and hepatoblastoma. J. Hepatol. 25, 445-453.
- Nishigori, H., Yamada, S., Kohama, T., Tomura, H., Sho, K., Horikawa, Y., Bell, G. I., Takeuchi, T., and Takeda, J. (1998). Frameshift mutation, A263fsinsGG, in the hepatocyte nuclear factor-1beta gene associated with diabetes and renal dysfunction. Diabetes 47, 1354-1355.
- Notenboom, R. G., de Boer, P. A., Moorman, A. F., and Lamers, W. H. (1996). The establishment of the hepatic architecture is a prerequisite for the development of a lobular pattern of gene expression. Development 122, 321-332.
- Nowell, P. C. (2002). Tumor progression: a brief historical perspective. Semin. Cancer Biol. 12, 261-266.
- Oda, H., Nozawa, K., Hitomi, Y., and Kakinuma, A. (1995). Laminin-rich extracellular matrix maintains high level of hepatocyte nuclear factor 4 in rat hepatocyte culture. Biochem. Biophys. Res. Commun. 212, 800-805.
- Odom, D. T., Zizlsperger, N., Gordon, D. B., Bell, G. W., Rinaldi, N. J., Murray, H. L., Volkert, T. L., Schreiber, J., Rolfe, P. A, Gifford, D. K, Fraenkel, E., Bell, G. I., and Young, R. A. (2004). Control of pancreas and liver gene expression by HNF transcription factors. Science 303, 1378-1381.
- Osanai, M., Chiba, H., Kojima, T., Fujibe, M., Kuwahara, K., Kimura, H., Satoh, M., and Sawada, N. (2002). Hepatocyte nuclear factor (HNF)-4alpha induces expression of endothelial Fas ligand (FasL) to prevent cancer cell transmigration: a novel defense mechanism of endothelium against cancer metastasis. Jpn. J. Cancer Res. 93, 532-541.
- Ossipow, V., Descombes, P., and Schibler, U. (1993). CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc. Natl. Acad. Sci. USA 90, 8219-8223.
- Ott, M.-O., Rey-Campos, J., Cereghini, S., and Yaniv, M. (1991). vHNF-1 is expressed in epithelial cells of distinct embryonic origin during development and precedes HNF-1 expression. Mech. Devel. 36, 47-58.
- Oya, M., Horiguchi, A., Mizuno, R., Marumo, K., and Murai, M. (2003). Increased activation of CCAAT/enhancer binding protein-beta correlates with the invasiveness of renal cell carcinoma. Clin. Cancer Res. 9, 1021-1027.
- Parviz, F., Matullo, C., Garrison, W. D., Savatski, L., Adamson, J. W., Ning, G., Kaestner, K. H., Zaret, K. S., and Duncan, S. A. (2003). Hepatocyte nuclear factor 4 alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 34, 292-296.
- Peinado, H., Portillo, F., and Cano, A. (2004). Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 48, 365-375.
- Perl, A. K., Welgenbus, P., Dahl, U., Semb, H., and Christofori, G. (1998). A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392, 190-193.
- Piechocki, M. P., Toti, R. M., Fernstrom, M. J., Burk, R. D., and Ruch, R. J. (2000). Liver cell-specific transcriptional regulation of connexin32. Biochim. Biophys. Acta. 1491, 107-122.
- Pontoglio, M., Barra, J., Hadchouel, M., Doyen, A., Babinet, C., and Yaniv, M. (1996). Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell 84, 575-585.
- Pontoglio, M., Sreenan, S., Roe, M., Pugh, W., Ostrega, D., Doyen, A., Pick, A. J., Baldwin, A., Velho, G., Froguel, P., Levisetti, M., Bonner-Weir, S., Bell, G. I., Yaniv, M., and Polonsky, K. S. (1998). Defective insulin secretion in hepatocyte nuclear factor 1 alpha-deficient mice. J. Clin. Invest. 101, 2215-2222.
- Pontoglio, M., Prie, D., Cheret, C., Doyen, A., Leroy, C., Froguel, P., Velho, G., Yaniv, M., and Friedlander, G. (2000). HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep. 1, 359-365.
- Potter, V. P. (1978). Phenotypic diversity in experimental hepatomas: The concept of partially blocked ontogeny. Br. J. Cancer 38, 1-23.
- Pupa, S. M., Menard, S., Forti, S., and Tagliabue, E. (2002). New insights into the role of extracellular matrix during tumor onset and progression. J. Cell Physiol. 192, 259-267.
- Radisky, D., and Bissel, M. J. (2004). Respect the neighbor. Science 303, 775-777.
- Radisky, D., Hagios, C., and Bissell, M. (2000). Tumors are unique organs defined by abnormal signaling and contact. Semin. Cancer Biol. 11, 87-95.
- Ramji, D. P., and Foka, P. (2002). CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365, 561-575.
- Rana, B., Xie, Y., Mischoulon, D., Bucher, N. L., and Farmer, S. R. (1995). The DNA binding activity of C/EBP transcription factor is regulated in the G1 phase of the hepatocyte cell cycle. J. Biol. Chem. 270, 18123-18132.
- Rask, K., Thorn, M., Ponten, F., Kraaz, W., Sundfeldt, K., Hedin, L., and Enerback, S. (2000). Increased expression of the transcription factors CCAAT-enhancer binding protein-beta (C/EBP Beta) and C/EBP zeta (CHOP) correlate with invasiveness of human colorectal cancer. Int. J. Cancer 86, 337-343.
- Rebouissou, S., Vasiliu, V., Thomas, C., Bellanne-Chantelot, C., Bui, H., Chretien, Y., Timsit, J., Rosty, C., Laurent-Puig, P., Chauveau, D., and Zucman-Rossi, J. (2005). Germline hepatocyte nuclear factor 1alpha and 1beta mutations in renal cell carcinomas. Hum. Mol. Genet. 14, 603-614.
- Rey-Campos, J., Chouard, T., Yaniv, M., and Cereghini, S. (1991). vHNF1 is a homeodomain that activates transcription and forms heterodimers with HNF1. EMBO J., 10, 1445-1457.
- Robinson, G. W., Johnson, P. F., Hennighausen, L., and Sterneck, E. (1998). The C/EBPb transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev. 12, 1907-1916.
- Runge, D., Runge, D. M., Bowen, W. C., Locker, J., and Michalopoulos, G. K. (1997). Matrix induced re-differentiation of cultured rat hepatocytes and changes of CCAAT/enhancer binding proteins. Biol. Chem. 378, 873-881.
- Runge, D., Runge, D. M., Drenning, S. D., Bowen, W. C., Jr., Grandis, J. R., and Michalopoulos, G. K. (1998). Growth and differentiation of rat hepatocytes: changes in transcription factors HNF-3, HNF-4, STAT-3, and STAT-5. Biochem. Biophys. Res. Commun. 250, 762-768.
- Runge, D., Runge, D. M., Daskalakis, N., Lubecki, K. A., Bowen, W. C., and Michalopoulos, G. K. (1999). Matrix-mediated changes in the expression of HNF-4alpha isoforms and in DNA-binding activity of ARP-1 in primary cultures of rat hepatocytes. Biochem. Biophys. Res. Commun. 259, 651-655.
- Ruoslahti, E. (1999). Fibronectin and its integrin receptors in cancer. Adv. Cancer Res. 76, 1-20.
- Ryffel, G. U. (2001). Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor HNF1 and HNF4 families: functional and pathological consequences. J. Mol. Endocrinol. 27, 11-29.
- Samadani, U., and Costa, R. H. (1996). The transcriptional activator hepatocyte nuclear factor 6 regulates liver gene expression. Mol. Cell. Biol. 16, 6273-6284.
- Sanchez, A., Alvarez, A. M., Lopez Pedrosa, J. M., Roncero, C., Benito, M., and Fabregat, I. (1999). Apoptotic response to TGF-beta in fetal hepatocytes depends upon their state of differentiation. Exp. Cell Res. 252, 281-291.
- Schmeichel, K. L., and Bissell, M. J. (2003). Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 116, 2377-2388.
- Schrem, H., Klempnauer, J., and Borlak, J. (2002). Liver-enriched transcription factors in liver function and development. Part I: The hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol. Rev. 54, 129-158.
- Schrem, H., Klempnauer, J., and Borlak, J. (2004). Liver-enriched transcription factors in liver function and development. Part II: the C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol. Rev. 56, 291-330.
- Seagroves, T. N., Krnacik, S., Raught, B., Gay, J., Burgess-Beusse, B., Darlington, G. J., and Rosen, J. M. (1998). C/EBPbeta, but not C/EBPalpha, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev. 12, 1917-1928.
- Sel, S., Ebert, T., Ryffel, G. U., and Drewes, T. (1996). Human renal cell carcinogenesis is accompanied by a coordinate loss of the tissue specific transcription factors HNF4 alpha and HNF1 alpha. Cancer Lett. 101, 205-210.
- Shen, C. N., Slack, J. M., and Tosh, D. (2000). Molecular basis of transdifferentiation of pancreas to liver. Nat. Cell. Biol. 12, 879-887.
- Shim, M., Powers, K. L., Ewing, S. J., Zhu, S., and Smart, R. C. (2005). Diminished expression of C/EBP alpha in skin carcinomas is linked to oncogenic Ras and reexpression of C/EBP alpha in carcinoma cells inhibits proliferation. Cancer Res. 65, 861-867.
- Slack, J. M. (1995). Developmental biology of the pancreas. Development 121, 1569-1580.
- Sladek, F. M., and Seidel, S. D. (2001). Hepatocyte nuclear factor 4a. In "Nuclear Receptors and Genetic Diseases" (Burris, T. P., and McCabe, E., Eds.), pp. 309-361. Academic Press, London.
- Sladek, F. M., Zhong, W., Lai, E., and Darnell, J. E., Jr. (1990). Liver-enriched transcription factor HNF4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 4, 2353-2364.
- Soriano, H. E., Kang, D. C., Finegold, M. J., Hicks, M. J., Wang, N. D., Harrison, W., and Darlington, G. J. (1998). Lack of C/EBP alpha gene expression results in increased DNA synthesis and an increased frequency of immortalization of freshly isolated mice hepatocytes. Hepatology 27, 392-401.
- Soutoglou, E., Papafotiou, G., Kartakili, N., and Talianidis, I. (2000). Transcriptional activation by hepatocyte nuclear factor-1 requires synergism multiple coactivator proteins. J. Biol. Chem. 275, 12515-12520.
- Spath, G. F., and Weiss, M. C. (1998). Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J. Cell Biol. 140, 935-946.
- Spear, B. T. (1999). Alpha-fetoprotein gene regulation: lessons from transgenic mice. Semin. Cancer Biol. 9, 109-116.
- Stahl, P. J., and Felsen, D. (2003). Transforming growth factor-beta, basement membrane, and epithelial-mesenchymal transdifferentiation. Am. J. Pathol. 159, 1187-1192.
- Stamenkovic, I. (2003). Extracellular matrix remodeling: the role of matrix metalloproteinases. J. Pathology 200, 448-464.
- Stenman, U. H., Leinonen, J., Zhang, W.-M., and Finne, P. (1999). Prostate-specific antigen. Semin. Cancer Biol. 9, 83-92.
- Stetler-Stevenson, W., and Yu, A. E. (2001). Proteases in invasion: matrix metalloproteinases. Semin. Cancer Biol. 11, 143-152.
- Stevenson, G. T., and Cragg, M. S. (1969). Molecular markers of B-cell lymphoma. Semin. Cancer Biol. 9, 139-147.
- Strathdee, G. (2002). Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin. Cancer Biol. 12, 373-379.
- Sugimoto, S., Mitaka, T., Ikeda, S., Harada, K., Ikai, I., Yamaoka, Y., and Mochizuki, Y. (2002). Morphological changes induced by extracellular matrix are correlated with maturation of rat small hepatocytes. J. Cell. Biochem. 87, 16-28.
- Sund, N. J., Ang, S. L., Sackett, S. D., Shen, W., Daigle, N., Magnuson, M. A., and Kaestner, K. H. (2000). Hepatocyte nuclear factor 3beta (Foxa2) is dispensable for maintaining the differentiated state of the adult hepatocyte. Mol. Cell. Biol. 20, 5175-5183.
- Sund, N. J., Vatamaniuk, M. Z., Casey, M., Ang, S. L., Magnuson, M. A., Stoffers, D. A., Matschinsky, F. M., and Kaestner, K. H. (2001). Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev. 15, 1706-1715.
- Sundfeldt, K., Ivarsson, K., Carlsson, M., Enerback, S., Janson, P. O., Brannstrom, M., and Hedin, L. (1999). The expression of CCAAT/enhancer binding protein (C/EBP) in the human ovary in vivo: specific increase in C/EBPbeta during epithelial tumour progression. Br. J. Cancer 79, 1240-1248.
- Suzuki, A., Iwama, A., Miyashita, H., Nakauchi, H., and Taniguchi, H. (2003). Role for growth factors and extracellular matrix in controlling differentiation of prospectively isolated hepatic stem cells. Development 130, 2513-2524.
- Taraviras, S., Monaghan, A. P., Schutz, G., and Kelsey, G. (1994). Characterization of mouse HNF-4 gene and its expression during mouse embryogenesis. Mech. Devel. 48, 67-79.
- Tenen, D. G. (2003). Disruption of differentiation in human cancer: AML shows the way. Nat. Rev. Cancer 3, 89-101.
- Thiery, J. P. (2002). Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442-454.
- Thomas, H., Jaschkowitz, K., Bulman, M., Frayling, T. M., Mitchell, S. M., Roosen, S., Lingott-Frieg, A., Tack, C. J., Ellard, S., Ryffel, G. U., and Hattersley, A. T. (2001). A distant upstream promoter of the HNF-4alpha gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum. Mol. Genet. 10, 2089-2097.
- Thorgeirsson, S. S., and Grisham, J. W. (2002). Molecular pathogenesis of human hepatocellular carcinoma. Nature Gen. 31, 339-346.
- Tirona, R. G., and Kim, R. B. (2005). Nuclear receptors and drug disposition gene regulation. J. Pharm. Sci. 94, 1169-1186.
- Torres-Padilla, M. E., Fougere-Deschatrette, C., and Weiss, M. C. (2001). Expression of HNF4alpha isoforms in mouse liver development is regulated by sequential promoter usage and constitutive 3' end splicing. Mech. Dev. 109, 183-193.
- Torres-Padilla, M. E., Sladek, F. M., and Weiss, M. C. (2002). Developmentally regulated N-terminal variants of the nuclear receptor hepatocyte nuclear factor 4alpha mediate multiple interactions through coactivator and corepressor-histone deacetylase complexes. J. Biol. Chem. 277, 44677-44687.
- Tronche, F., and Yaniv, M. (1992). HNF-1, a homeoprotein member of the hepatic transcription regulatory network. Bioessays 14, 579-587.
- Tronche, F., Ringeisen, F., Blumenfeld, M., Yaniv, M., and Pontoglio, M. (1997). Analysis of the distribution of binding sites for a tissue-specific transcription factor in the vertebrate genome. J. Mol. Biol. 266, 231-245.
- Tsuchiya, A., Sakamoto, M., Yasuda, J., Chuma, M., Ohta, T., Ohki, M., Yasugi, T., Taketani, Y., and Hirohashi, S. (2003). Expression profiling in ovarian clear cell carcinoma: identification of hepatocyte nuclear factor-1 beta as a molecular marker and a possible molecular target for therapy of ovarian clear cell carcinoma. Am. J. Pathol. 163, 2503-2512.
- Vaccariello, M., Javaherian, A., Wang, Y., Fusenig, N. E., and Garlick, J. A. (1999). Cell interactions control the fate of malignant keratinocytes in an organotypic model of early neoplasia. J. Invest. Dermatol. 113, 384-391.
- Valdes, F., Alvarez, A. M., Locascio, A., Vega, S., Herrera, B., Fernandez, M., Benito, M., Nieto, M. A., and Fabregat, I. (2002). The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol. Cancer Res. 1, 68-78.
- Van Dongen, J. J., Szczepanski, T., and Adriaansen, H. J. (2002). Immunology of leukemia. In "Leukemia" (E. Henderson, T. A. Lister, and M. F. Greaves, Eds.), 7 Edition, pp. 85-129. Sanders, Philadelphia.
- Varga, E. V., Cheremnova, O. A., Ovchinnikov, D. A., Shapiguzov, A. Yu., Kudryavtseva, E. I., Morozova, O. V., Engelhardt, N. V., and Lazarevich, N. L. (2001). The tissue-specific genes expression during murine hepatocarcinoma progression. Genetika (Russ). 37, 803-810.
- Wan, H., Dingle, S., Xu, Y., Besnard, V., Kaestner, K. H., Ang, S.-L., Wert, S., Stahlman, M. T., and Whitsett, J. A. (2005). Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J. Biol. Chem. 280, 13809-13816.
- Wang, L., Coffinier, C., Thomas, M. K., Gresh, L., Eddu, G., Manor, T., Levitsky, L. L., Yaniv, M., and Rhoads, D. B. (2004). Selective deletion of the Hnf1beta (MODY5) gene in beta-cells leads to altered gene expression and defective insulin release. Endocrinology 145, 3941-3949.
- Wang, N. D., Finegold, M. J., Bradley, A., Ou, C. N., Abdelsayed, S. V., Wilde, M. D., Taylor, L. R., Wilson, D. R., and Darlington, G. J. (1995). Impaired energy homeostasis in C/EBP alpha knockout mice. Science 269, 1108-1112.
- Wang, W., Hayashi, Y., Ninomiya, T., Ohta, K., Nakabayashi, H., Tamaoki, T., and Itoh, H. (1998). Expression of HNF-1 alpha and HNF-1 beta in various histological differentiations of hepatocellular carcinoma. J. Pathol. 184, 272-278.
- Watkins, P. J., Condreay, J. P., Huber, B. E., Jacobs, S. J., and Adams, D. J. (1996). Impaired proliferation and tumorigenicity induced by CCAAT/enhancer-binding protein. Cancer Res. 56, 1063-1067.
- Weaver, V., Lelievre, S., Lakins, J., Chrenek, M., Jones, J., Giancotti, F., Werb, Z., and Bissell, M. (2002). β4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2, 205-216.
- Weaver, V., Petersen, O. W., Wang, F., Larabell, C., Briand, P., Damsky, C., and Bissell, M. J. (1997). Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 137, 231-245.
- Weinberg, R. A. (1989). Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res. 49, 3713-3721.
- Weinstein, D. C., Ruiz i Altaba, A., Chen, W. S., Hoodless, P., Prezioso, V. R., Jessell, T. M. and Darnell, J. E., Jr. (1994). The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell 78, 575-588.
- Xu, L., Hui, L., Wang, S., Gong, J., Jin, Y., Wang, Y., Ji, Y., Wu, X., Han, Z., and Hu, G. (2001). Expression profiling suggested a regulatory role of liver-enriched transcription factors in human hepatocellular carcinoma. Cancer Res. 61, 3176-3181.
- Yamada, K. M., Pankov, R., and Cukierman, E. (2003). Dimensions and dynamics in integrin function. Braz. J. Med. Biol. Res. 36, 959-966.
- Yamagata, K., Nammo, T., Moriwaki, M., Ihara, A., Iizuka, K., Yang, Q., Satoh, T., Li, M., Uenaka, R., Okita, K., Iwahashi, H., Zhu, Q., Cao, Y., Imagawa, A., Tochino, Y., Hanafusa, T., Miyagawa, J., and Matsuzawa, Y. (2002). Overexpression of dominant-negative mutant hepatocyte nuclear fctor-1 alpha in pancreatic beta-cells causes abnormal islet architecture with decreased expression of E-cadherin, reduced beta-cell proliferation, and diabetes. Diabetes 51, 114-123.
- Yamasaki, H. (1990). Gap junction intercellular communication and carcinogenesis. Carcinogenesis. 7, 1051-1058.
- Yano, T., Kishimoto, T., Tomaru, U., Kawarada, Y., Kato, H., Yoshiki, T., and Ishikura, H. (2003). Further evidence of hepatic transdifferentiation in hepatoid adenocarcinomas of the stomach: quantitative analysis of mRNA for albumin and hepatocyte nuclear factor-4alpha. Pathology 35, 75-78.
- Zahnow, C. A., Cardiff, R. D., Laucirica, R., Medina, D., and Rosen, J. M. (2001). A role for CCAAT/Enhancer binding protein b-liver-enriched inhibitory protein in mammary epithelial cell proliferation. Cancer Res. 61, 261-269.
- Zahnow, C. A., Younes, P., Laucirica, R., and Rosen, J. M. (1997). Overexpression of C/EBPbeta-LIP, a naturally occurring, dominant-negative transcription factor, in human breast cancer. J. Natl. Cancer Inst. 89, 1887-1891.
- Zaret, K. S. (2002). Regulatory phases of early liver development: paradigms of organogenesis. Nat. Rev. Genet. 3, 499-512.
- Zavadil, J., Bitzer, M., Liang, D., Yang, Y-C., Massimi, A., Kneitz, S., Piek, E., and Böttinger, E. P. (2001). Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad. Sci. USA 98, 6686-6691.
- Zhu, S., Oh, H. S., Shim, M., Sterneck, E., Johnson, P. F., and Smart, R. C. (1999). C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol. Cell. Biol. 19, 7181-7190.
- Zhu, S., Yoon, K., Sterneck, E., Johnson P. F., and Smart, R. C. (2002). CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. USA 99, 207-212.