| Home page (in Russian) | Texts in English | Guestbook |
Oncodevelopmental Biology and Medicine, 4 (1983) 371-381
Elsevier
Laboratory of Tumor Immunochemistry, Cancer Research Center, U.S.S.R.
Academy of Medical Sciences, Moscow, U.S.S.R.
(Accepted 14 April, 1983)
A historical account of the finding of alphafetoprotein is presented.
The problems in its initial isolation, purification and source are described.
Some interesting potential future studies are also discussed.
| Alphafetoprotein Hepatoma | Germ cell tumor Oncofetal antigen |
There was nothing unexpected in the finding that tumors can synthesize embryonic antigens. Nevertheless, the finding that embryonal serum protein was produced by liver carcinomas was a great surprise to us. Here is how it all happened.
Soon after graduating from Moscow University, I started work in 1951 with Professor Lev Alexandrovich Zilber (1894–1966), an outstanding virologist and immunologist with a brightly gifted and attractive personality. At that time, Zilber was one of the few people in the world carrying out active cancer immunology investigations.
To approach the problem we used anaphylaxis with a desensitization method, developed by Zilber a few years earlier. Guinea pigs were sensitized with tumor extracts, desensitized with normal tissue extracts and, after complete desensitization, challenged with the original tumor preparation. The anaphylactoid shock obtained after challenge was attributed to antigen(s) present in the tumor but absent from the normal tissue [33].
These results suggested antigenic specificity for tumors, but it was extremely difficult, if at all possible, to identify by this method the individual antigen (or antigens) responsible for such specificity. However, in the 1950's there was no other way to approach the problem.
It was logical to begin with the isolation and comparison of subcellular fractions of tumor and normal tissue. I was concerned initially with these problems in Zilber's department. In the early fifties, Ouchterlony suggested his precipitation-in-gel technique and Grabar and Williams [36] developed immunoelectrophoresis. These methods permitted the resolving of individual antigens in complex mixtures and comparing of individual antigens, present in different systems, without their preliminary isolation and purification. Analytical biochemistists in those days could not even imagine anything of the kind, and I was deeply impressed by the exciting possibilities opened up by these methods. However, we thought then that such a rough and not very sensitive reaction as the precipitation test would hardly be able to reveal subtle antigenic distinctions between tumor and normal tissues.
Between 1956 and 1957, we started to use immunodiffusion to study tissue antigens. The liver and hepatomas of mice became an object of study. In cooperation with the biochemists Z.A. Avenirova and N.V. Engelhardt, who had just joined us after graduating from the University, we found that precipitation in gel clearly demonstrated loss of several liver-specific antigens from hepatomas. These antigens could be identified and characterized precisely, as well as be employed for reliable biochemical studies [7,–8].
In the same experiments we detected, though not very regularly, an antigen occurring in hepatomas, but absent from the liver. We were mainly interested in normal antigens lost by the tumor as, in this context, we were on safe technical ground. However, Zilber insisted that all our efforts should be directed towards identifying the hepatoma-associated antigen, undoubtedly more interesting but not very convenient for study.
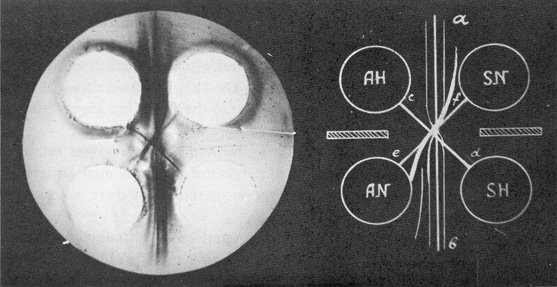
It was difficult to get to grips with the problem. Its fine precipitin line appeared irregularly in the complex spectrum of bands produced by the antigens of the liver and hepatomas. By changing the standard Ouchterlony arrangement of wells into a 'square pattern' [1], detection of the antigen became more reproducible.
Fig. 1. Comparison of normal liver and hepatoma extracts in 'square' pattern.
AH, hepatoma extract; AN, liver extract; SH and SN, antisera to hepatoma and liver, respectively;
a-b, precipitin lines of antigens shared by normal liver and hepatoma; c-d, liver-specific antigen; e-f, hepatoma-specific antigen.
(From Ref. 35)
In the summer of 1958 at the seventh International Cancer Congress in London, Zilber was able to demonstrate the hepatoma antigen (AH) which was absent from the liver (Fig. 1) [34, 35]. Using a simple test for AH, we started, in cooperation with V.S. Tsvetkov, its isolation from hepatoma extracts. Over the next 18 months we developed a new technique for antigen purification – 'immunofiltration' – and succeeded in preparing the antigen. In immunofiltration, we used the immunological specificity of AH for its purification. At that time, there were no antibody immunosorbents, so we employed free antibodies to normal antigens to separate them from the hepatoma-specific one.
First, the hepatoma extract was fractionated by ammonium sulfate. AH and serum albumin were, together with some other impurities, precipitated in this way. Next, we purified the rough fraction by preparative electrophoresis in agar gel and obtained an electrophoretically homogeneous fraction with the mobility of serum α-globulins and which contained AH as its dominant component (Fig. 2).
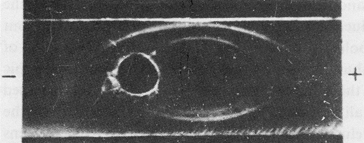
Fig. 2. Immunoelectrophoresis of the α-globulin fraction of a hepatoma extract.
Developed: upper trench, anti-hepatoma serum; lower trench, anti-liver serum. (From Ref. 14)
The immunofiltration technique was used to separate AH from antigens common to the liver. For this purpose an anti-liver serum was subjected to electrophoresis in agar gel block. Serum immunoglobulins moved towards the cathode by electroosmotic flow. When all immunoglobulins were included in the agar block, the entire area of the block initially orientated to the anode was removed and replaced by fresh agar.
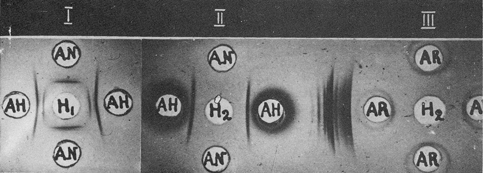
Fig. 3. Purification of hepatoma-specific antigen by immunofiltration.
I, α-Globulin fraction (H1) before immunofiltration; AH and AN, antisera to hepatoma and liver, respectively.
II, Purified antigen (H2) after immunofiltration.
III, Absence of rabbit serum proteins in purified fraction (H2).
AR, Donkey antiserum to rabbit serum proteins. (From Ref. 14)
Thus, all the serum proteins with anodic mobility in agar were withdrawn. Then, the AH-containing fraction was introduced into a fresh part of the agar block, just in front of the immunoglobulins, and the polarity was reversed. Countercurrent migration of AH to the anode and immunoglobulins towards the cathode took place. Antigenic admixtures were precipitated by the corresponding antibodies; AH freely passed through the 'antibody filter', while all unbound immunoglobulins were displaced to the cathodic part of the block. After elution from the agar, AH was free from antigens shared by the liver and hepatomas, as well as from the proteins of the antiserum used for immunofiltration (Figs. 3, 4).
Thereby, we isolated and purified AH, then considered by us to be the first tumor-specific antigen [14,15].
At the same time we obtained purified antibodies to AH by partial dissociation of its specific precipitate made by a hepatoma extract and antihepatoma serum, absorbed by normal antigens. These antibodies detected AH only in the hepatoma extract and did not react with normal mouse liver, kidney or spleen extracts (Fig. 5) [7]. Thus, we now had an absolutely reliable test to detect precisely and reproducibly the hepatoma antigen in any system. This antigen proved to be, indeed, specific. AH was missing in all the organs of normal mice, but it was found regularly, and in great amounts, in hepatoma extracts. We were able to offer the antigen to any laboratory and we were, naturally, proud of having been the first to isolate a tumor-specific antigen.
However, the nearer we approached our goal, the more we had doubts. Of these, there were three in particular.
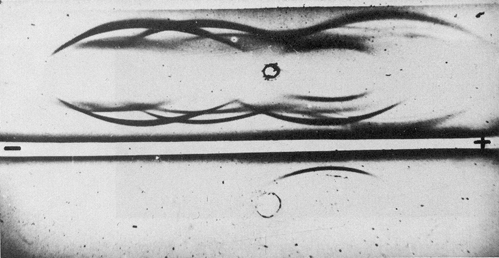
Fig. 4. Immunoelectrophoresis of purified hepatoma-specific antigen.
Upper well, hepatoma extract; bottom well, purified antigen.
Central trench, anti-hepatoma serum; peripheral trenches, anti-liver serum.
First, our antigen might possibly be an antigen of a passenger virus, having contaminated the transplantable hepatoma strain. (The hepatoma XXII strain was originally produced by V.I. Gelstein in 1951 by transplanting hepatoma of C3HA mouse fed with ortho-aminoarotoluene.) One case supported this possibility. Once, for a control we got from another Institute a transplantable gastric carcinoma and, surprisingly, found AH in it. Fortunately, the 'stomach cancer' proved to be contaminated by our hepatoma, which replaced the original strain, and AH had enabled us to reveal this. However, possibility of viral contamination in our strain still could not be ruled out altogether.

Fig. 5. Purified antibodies to liver and hepatoma-specific antigens.
eH, antibody to hepatoma-specific antigen; eN, antibody to liver-specific antigen;
N and H, extracts from the liver and hepatoma, respectively;
AH, anti-hepatoma serum. (From Ref. 7)
The second problem was even more important. Our hepatoma strain differed sharply in morphology and antigenic composition from the normal liver. While it had originated in liver, what cell type was responsible for tumor formation? If it originated from bile duct epithelia, rather than from hepatocytes, there was nothing surprising in the fact that the tumor did not contain liver-specific antigens, but possessed its 'own' antigen, not tumor-specific but specific for the biliary epithelium.
In that case, the significance of AH would be considerably decreased. Our antigen would belong to a quite common group of tissue-specific antigens and not be a tumor-specific one. At that time, it was very difficult to exclude this possibility. Immunofluorescence techniques were practically absent from our country. Fluorescent dyes, fluorescence microscopes and cryostats were not available, and nobody had any experience of using immunofluorescence on tissue sections. Besides, AH, as a secreted protein, proved to present many difficulties in its localization at the tissue level, and it was not until 1969 that the first reliable results were obtained [21, 24]. However, we immediately started immunofluorescent studies of AH and other liver antigens as soon as we realized the necessity of elucidating which cell type was responsible for giving rise to the hepatoma strain used in our investigations.
Finally, the most trivial possibility was that our antigen was present in the normal liver, but in subthreshold levels not detectable by immunodiffusion methods. Thus, we also had to develop more sensitive assays for AH.
In 1960–1961, we attempted to solve all these problems. Surprisingly, the study acquired quite another direction.
In parallel with the AH studies, we continued our investigations on the liver-specific antigens and their loss in liver tumors, performing this work together with a young colleague from our group – Ninel Khramkova (now N.I. Kuprima). The study was successful and we obtained test systems for the detection of several liver-specific antigens. Different transplantable hepatomas were used in the study and each hepatoma strain was shown to have its own individual pattern of liver-specific antigens [3]. This might imply that each hepatoma lost some particular antigens due to malignant transformation or tumor progression, but it could also mean that different hepatomas corresponded to different stages of liver development with their own peculiar and/or incomplete sets of tissue-specific antigens.
To test this possibility we started to study 'antigenic maturation' of murine livers in the early stages of development. Such work was safe in technical terms, so we decided to involve a medical school student who was doing her training in our laboratory at that time. AH was included in these experiments as an additional control of its tumor specificity. Our student began the work! The next day she did not come to look at her results. We have never seen her again. When we examined her results we could hardly believe our eyes. Enormous amounts of AH were present in the liver of the mouse embryo! Never before had we seen such amounts of AH in any system and we could not even guess what mistake the student had made to obtain this result. The experiment was immediately repeated with due care and every precaution. The results were the same! Then, we included in the experiment other embryonic organs – spleen, kidney, heart, brain. The results was identical – an enormous amount of the antigen was observed in all the organs.
Most probably the antigen arose from serum rather than from tissue, and penetrated different organs with the blood. Accordingly, embryonic blood was examined and the 'hepatoma antigen' was detected there in dilutions of up to 1:4000, much higher than in various organ extracts, including the liver.
Thus, surprisingly, AH appeared to be a serum antigen instead of tumor– or even tissue-associated one. Its appearance in the tumor, therefore, might be due simply to blood contamination.
Immediately we tested the blood of hepatoma-bearing mice and, indeed, found AH there in higher quantities than in the tumor tissue itself, which we had always tried to wash out of the blood very carefully. AH also proved to occur in different organs of hepatoma-bearing mice.
So we began to feel uncertain: AH penetrated the tumor with the blood and, quite possibly, bore no relation to hepatomas being produced elsewhere as a response to the tumor graft. In this way, it could be similar to an acute phase protein, and could be transported to the tumor and deposited there in its hemorrhagic areas and possess nothing in common with a tumor-specific antigen. Such a simple interpretation seemed quite plausible. After all, a rat serum protein had already been described which had similar properties to AH, was also an α-globulin and was known to occur in the sera of tumor-bearing animals [17, 18].
I was almost sure that this was the case. Immediately we obtained from the laboratory several mice grafted with different sarcomas and examined their sera for AH. One was positive. Our worst assumption seemed to be confirmed. At this point there remained only a faint glimmer of hope. Perhaps the AH-positive mice might be pregnant. Keeping in mind the enormous AH level in fetal serum, it was natural to expect small amounts of the antigen in maternal blood. In the cage from which we had taken the sarcoma-bearing animals there were, indeed, newborn mice. But which mouse had borne them?
Next time, we took blood only from males bearing sarcomas and other non-liver tumors, and, separately, the blood of pregnant females. AH was found only in pregnant females. There was no AH in animals with non-liver tumors. Now, we felt somewhat easier. However, the site of synthesis of the 'hepatoma antigen' remained unknown. It might be the liver for instance, which could produce AH only in response to a hepatoma. Without knowing the origin of the antigen, our data could not be interpreted properly.
That was a difficult, but soluble, problem. In cooperation with our biologically orientated colleagues we started three sets of experiments: (1) the heterotransplantation of mouse hepatomas into the hamster cheek pouch; (2) the heterotransplantation of mouse hepatomas to cortisone-treated rats; and (3) explanting hepatomas into tissue culture.
In all three systems, there was no response of the murine organism to the growth of the tumor.
These experiments were successful. The small tumor nodules in the hamster cheek pouch were found to have some AH in them. However, it was difficult to be sure that this AH was not present as a 'contaminant' in the grafted tissue.
Rapid growth of the hepatoma was observed in cortisone-treated rats. AH accumulated in their sera and the dynamics of its rise and fall corresponded to the growth and subsequent regression of the tumor. However, these experiments did not seem to us to be absolutely conclusive, since the rats may well produce their own AH indistinguishable from the murine one, synthesized in response to mouse hepatoma. This possibility had to be excluded before any final interpretation of the heterotransplantation experiments could be made.
In trying to solve this problem, we have found that the rat can produce its own 'AH', which is actually analogous, but not identical, to murine AH. It cross-reacted with the murine antigen, but definitely differed from the latter in its immunological properties. In these first experiments, we used the immunodiffusion test system to murine AH. The antiserum was not very strong and rat 'AH' did not form a precipitin band with it, but inhibited precipitation with a homologous antigen. The inhibition activity was strictly specific. According to the inhibition titre, we estimated the amount of rat 'AH' in different systems. Later, we obtained antisera to rat embryonic α-globulin and demonstrated its partial identity to the murine one [3].
As in mice, rat 'AH' was detected in high amounts in the blood of rat embryos, was missing in the blood of adults, but reappeared in the serum and ascitic fluid of adult rats grafted with the Zajdela rat hepatoma. These experiments were quite conclusive and clearly demonstrated that AH is produced by the hepatoma, and then secreted into the blood.
By then, primary cultures of our hepatomas had been established. These cultures accumulated AH in the medium, which was changed several times during culture. Besides AH, the cultures also contained in the medium other serum proteins – albumin and transferrin secreted by the tumor cells.
We had already obtained antisera to murine embryonic serum proteins. After absorption with adult murine serum these antisera revealed AH very distinctly. The serum and hepatoma antigens were completely identical, in both their physicochemical and antigenic properties.
Now we could dot the 'i's' and cross the 't's'. It became clear that AH was a normal component of embryonic serum (subsequently we referred to it as the αF-globulin, i.e., fetal α-globulin*, which is most likely to be synthesized by the fetal liver, where the majority of serum proteins are synthesized**. Its production ceased a month after birth and was resumed only in liver carcinomas, which are responsible for its production and secretion into the blood. There was a wide range of aF production by hepatomas, being especially pronounced in poorly differentiated tumors. Low level resumption of its synthesis could be induced temporarily in adult mice by partial hepatectomy. It seemed to us most likely that aF was a specific product of proliferating hepatocytes, thereby accounting for its synthesis by hepatomas.
|
* In 1969, a WHO expert group introduced the term α-fetoprotein (AFP) for this protein. It was clear at that time that AFP was first found in 1956 in human serum by Bergstrand and Czar (for review, see Ref. 4). |
All these experiments, from the first detection of AH in the embryo in January 1962 to the demonstration of its synthesis in tissue culture, took little more than 4 months. The situation became even more complicated because in July 1962 in Moscow the eighth International Cancer Congress was to be held, and Professor P. Grabar had invited me to deliver a communication on our studies at the panel discussion on tumor antigens. The abstract had been submitted a long time ago. Professor Grabar knew of our previous results on the hepatoma-associated antigen and was interested in them. In July we were expected to give an answer as to the nature of the hepatoma antigen. Time was short.
Many guests were expected to take part in the Congress. The Gamaleya Institute, in making preparations for their reception, had been repairing our laboratory since the early spring. Rooms were painted, whitewashed and plastered. At any moment we could be asked to free the rooms; everywhere there was builders refuse, and even now I wonder how we managed to carry on with our work despite the turmoil around us.
In July 1962 our results on the production of embryonic α-globulin by hepatomas were reported at the eighth Cancer Congress, and then at the Soviet-French Symposium held in Moscow immediately after the Congress [2,11].
The results – a specific hepatoma-associated antigen proving also to be embryonic in nature – were greeted with a certain amount of surprise, as well as interest. In August, we submitted two full-length papers – one in Russian to Biokhimia [12] and the other to Transplantation [13]. Because of personal contacts at the Congress, and demonstrations in the laboratory, the results with αF quickly became known at home and abroad. 2 years later they received full confirmation in Villejuif in Grabar's laboratory and, later, in other laboratories.
At that time we were rather sceptical of using our findings in an immunodiagnostic context. First, one still had to show that αF-globulin was producted by human hepatomas, not least because we had found that not all primary hepatomas in mice produced this protein, while the regenerating liver resumed its synthesis. Accordingly, we expected αF-globulin production in different liver disorders to be accompanied by regeneration. An important immunodiagnostic step was taken next year by an Astrakhan biochemist, Yu.S. Tatarinov, who found embryo-specific α-globulin in the blood of a patient with primary liver cancer. Shortly after, Yu.S. Tatarinov already had under observation four hepatoma-bearing patients, containing αF-globulin in the blood, along with negative controls. He reported his observations to the first U.S.S.R. Biochemical Congress in January 1964 [31, 32].
Thus, the diagnostic aspect of the problem arose, and soon it became of great importance to both experimentalists and clinicians. However, I was sure that in man the case was actually much the same as in mice. For several years, in collaboration with A.M. Olovnikov, V.S. Tsvetkov and D.A. Elgort, we had been developing a highly sensitive test for the detection og AFP. Finally, we succeeded in evolving it on the basis of an immunoautoradiographic method [30], and AFP was also found to be actually synthesized by adult patients with acute viral hepatitis [16].
Among the very first papers devoted to human αF, one should specially mention the investigation carried out by some Prague biochemists [28], who demonstrated very high levels in a child with a hepatoblastoma.
Later, we also performed clinical studies in cooperation with physicians and pathologists from the Institute of Experimental and Clinical Oncology in Moscow. In these studies we confirmed, by means of vast amounts of clinical material, the diagnostic value of the αF-test in primary liver cancer, but also discovered its regular occurrence in the blood of patients with testicular germ cell tumors [6], in which AFP has proved to be a most valuable diagnostic marker, just as in hepatomas. Similar findings were independently reported by Masopust et al. [29], who also showed that serum AFP levels could reflect the efficacy of tumor treatment.
Since the first steps in the analysis of the AFP phenomenon, the cellular basis of its production seemed to me most important to understand, together with the rationale as to how this protein was regulated and reexpressed in tumors.
This approach revealed that the yolk sac endoderm was a second site of AFP production [25]. The finding of AFP in the histological structures of germ cell tumors similar to yolk sac visceral endoderm gave a rational explanation for its production by such tumors [23].
The demonstration of AFP in adult hepatocytes of regenerating liver, which did not enter S phase, showed that AFP synthesis was reversibly repressed in the mature hepatocytes [22].
Immunomorphological studies on the developing liver led us to conclude that liver plate formation is the critical event resulting in reversible repression of AFP in hepatocytes. Reappearance of AFP in a few cells surrounding the necrotic area in toxin-injured liver suggests that liver plate disarrangement in those areas is responsible for AFP reexpression by mature hepatocytes [4, 5].
Elegant studies by Dziadek and Adamson [20] and Dziadek [19] clearly demonstrated that contacts of the visceral endoderm and extraembryonic ectoderm lead to AFP expression both in vivo and in vitro.
In hepatic carcinogenesis, a cellular approach has shown transitory forms of hepatocytes as the source of AFP in the acute phase of chemical carcinogenesis [26, 27].
Thus a number of new, essentially unexpected and complicated problems have been created. Shall we have the strength and luck to contribute to their solution?