Методы всю жизнь имели для меня совершенно особое значение. Не потому, что у меня были какие-либо технические склонности, но как аппарат проникновения в природу явлений и как почва под ногами – зыбкая или надежная.
Я поступил на Биофак МГУ, чтобы изучать «природу мысли и сознания». На 2-м курсе (1946 г.) мы с моим, ныне покойным, товарищем Сашей Зотиным организовали кружок биофизики, хотя тогда такой науки не было. Мы подразумевали под ней некую «биомеханизмику» – науку о механизмах биологических процессов, в том числе (для меня) нервного процесса. От Саши я узнал про А.Г. Гурвича (так называемого «старого Гурвича» в отличие от иммунолога А.Е. Гурвича), про митогенетические лучи и теорию биологического поля. Митогенетические лучи привлекали меня как индикаторы специфичности нервного процесса – прохождения импульса по нерву. Я все прочитал про это и вместе еще с тремя студентками из кружка стал ходить и работать в лабораторию А.Г. Гурвича – на Балтийском поселке, тогдашней темной окраине Москвы, в старом Институте экспериментальной медицины. С нами занималась дочь А.Г., Анна Александровна, очень серьезная, доброжелательная и преданная сотрудница А.Г. Все было бы очень хорошо, но биологическая детекция излучения у меня не получилась. Митогенетические лучи определялись по индукции почкообразования у дрожжей. Число «почек» на 10³ клеток было показателем эффекта. Превышение над контролем ~ на 20% считалось хорошим эффектом. Вся работа сводилась к небольшому пробирочному эксперименту и бесконечному счету. Но счет, – до отчаяния, месяцами – не получался. Только-только что-то намечалось и вновь рушилось. Почвы под ногами не было. Анна Александровна старалась помочь, но – тщетно! Я терял уверенность в себе и в своих способностях к научной работе. Во время этой работы я сделал маленькое усовершенствование – пробирку с раствором, который облучался ультрафиолетом в одном месте, а индуцированное им митогенетическое излучение регистрировалось в другом, не облученном ультрафиолетом. И придумал и сделал этот сосуд я сам. Это мне так понравилось и дало ощущение такой простоты, ясности и твердости, что меня уже навсегда потянуло к таким вещам.
Я работал в лаборатории А.Г. года полтора и на середине третьего курса, скорее по интуиции и тяге к пробирочному эксперименту, пошел к А.Н. Белозерскому на кафедру биохимии растений. Конечно, даже и для себя я старался подвести под этот крутой поворот (нейрофизиология – биохимия животных – биохимия растений) солидную и убедительную теоретическую базу. Но простота биохимии (кажущаяся!) вместе с простотой растительных объектов и отсутствием вивесекций, которые я проделывал, преодолевая себя, сыграли свою, скорее подсознательную, роль. Приход на кафедру и работу на дипломе у Белозерского я описал в воспоминаниях об Андрее Николаевиче (Абелев, 1995; гл. II первой части этой книги).
Методы всегда имели для меня особую притягательность. Только те проблемы, к которым не было готовых методических подходов, казались мне по-настоящему интересными и стоящими. Проблемы, еще не решенные, но обеспеченные методически, представлялись уже принципиально решенными и лишь оставленными для доработки в случае свободного времени или наличия каких-либо дополнительных возможностей. При этом меня привлекала не техническая сторона метода, а его разрешающая способность, возможность расчленить сложный многокомпонентный или чисто эмпирический феномен на составляющие, понятные или элементарные компоненты, т.е. решить изучаемую задачу. Разработать или найти разрешающий метод казалось мне всегда более важным и интересным, чем даже получить искомый результат. Работа «по протоколам» (заранее отработанным и детально описанным методам) казалась мне всегда абсурдной и неинтересной, хотя она является одной из причин высочайшей продуктивности американской науки (1) Аналитические задачи были постоянными спутниками нашей работы, и сам я стремился сосредоточаться именно на них, зачастую оставляя получение результатов сотрудникам. Интерес к методам, их модификациям или разработкам стал складываться еще со студенческих времен и первым таким шагом была модификация метода раздельного определения ДНК и РНК по Шмидту и Тангаузеру (1945 г.) применительно к бактериальным объектам. Мой руководитель, проф. А.Н. Белозерский, предложил мне применить этот метод, разработанный авторами для анализа тканей животных, для бактериальных систем, и здесь метод оказался непригодным. Щелочной гидролизат бактериальной массы не разделялся осаждением HCl на ДНК- и РНК-содержащие фракции, и я долго бился, чтобы разделить их. Решение, в конце концов, было найдено, и А.Н. Белозерский включил мою модификацию в руководство по биохимии растений вместе с описанием получения гистонов (2) из растений, также сделанном в моем дипломе (см. Белозерский и Проскуряков, 1951).
Работа по гистонам меня глубоко увлекла уже по существу проблемы, – я был уверен, что гистоны должны быть не только в клетках эукариот, но и у бактерий. Попав волею судьбы и случая в биохимический отдел Института эпидемиологии и микробиологии им. Н.Ф. Гамалеи АМН СССР, и с благословения ныне покойного заведующего отделом Василия Андреевича Благовещенского (3), я стал дорабатывать свою модификацию метода Шмидта и Тангаузера и одновременно готовиться к выделению гипотетических бактериальных гистонов. В.А. организовывал тогда биохимический отдел, он был секретарем партбюро Института, постоянно занят, и был рад, что пришедший молодой биохимик сам чего-то хотел и знал, как к этому приступить. Он не только не возражал, но дал мне рабочий стол (целый прекрасный рабочий стол!) в своем кабинете и полный «карт-бланш» для работы. Был я тогда препаратором, даже не старшим лаборантом, и В.А. содействовал мне в получении подработки. Он ко мне вполне хорошо относился, и добрые отношения сохранились у нас на всю жизнь, несмотря на сложные, временами, ситуации, разводившие нас на противоположные позиции, например, в «деле Гурвича» (4).
Для изоляции гистонов у бактерий, – а это я хотел сделать по аналогии с гистонами растений, получив сначала «хромозин», структурированный ДНК-нуклеопротеид, – надо было мягко разрушить бактерии, что было нелегкой и очень специальной задачей. Большинство бактерий были чрезвычайно прочны механически (капсулы, бактериальные стенки), для разрушения их требовались специальные «пушки», основанные на разрыве бактерий при резком перепаде давления, или особые шаровые мельницы. Ни того, ни другого в институте не было, и тогда я решил использовать Micrococcus lysodeiticus, полисахаридная стенка которого специфически лизировалась ферментом лизоцимом.
С помощью лаборантов-микробиологов я нарастил много матрасов микрококков, получил из белка куриных яиц лизоцим и приготовил лизат бактериальных тел. Но лизат был столь вязким, что расфракционировать его в растворах NaCl разной концентрации, а это был основной способ отделения ДНК-протеидов от цитоплазматических белков, никак не получалось. Я уперся в тупик. А тут еще соседние микробиологи, обнаружив, что у них под носом выращиваются громадные количества посторонних и весьма агрессивных микрококков, начали косо смотреть на мои занятия и, наверное, жаловаться начальству. Работа моя забуксовала. При том еще обнаружилась все возраставшая у меня тяга к отделу Л.А. Зильбера, где главной проблемой были нуклеопротеиды опухолей, в которых Л.А. искал опухолеродный вирус и обнаруживал специфические опухолевые антигены, предположительно считавшиеся вирусными. А.Н. Белозерский консультировал эти работы. Я познакомился с биохимиками отдела – Зинаидой Алексеевной Авенировой и Валентиной Александровной Артамоновой. Мы хотели затеять совместную работу по фракционированию тотальной нуклеопротеидной фракции опухолей на ДНК- и РНК-протеидную. Однако, Благовещенский был резко против совместной работы с Зильбером. Он говорил, что союз с Зильбером – это «союз лошади и всадника». Но меня все более и более тянуло в этот отдел, где нуклеопротеиды были в центре интереса и внимания, а жизнь была напряженной и волнующей. Проблемы же отдела биохимии микробов – бактериальные антигены и токсины – были от меня очень далеки, а стиль работы – спокойный и неторопливый – был тоже мне не близок. Но все это происходило при хорошем и вполне терпимом ко мне отношении со стороны Благовещенского. Он даже дал мне препаратора в помощь – шуструю и бойкую девочку, с которой я не знал, что делать, т.к. на кафедре был приучен все делать сам и ничего, особенно мытья посуды, никому не доверять.
В это время (1951–1952 гг.) Благовещенскому предложили работу в закрытом бактериологическом институте в Загорске по бактериальным токсинам, а в отдел пришел В.С. Гостев – биохимик от Н.Н. Жукова-Вережникова (лысенковца от микробиологии и иммунологии). Это был человек неприятный, весьма серый, но амбициозный и люто ненавидящий Л.А. Зильбера. С сотрудниками же Зильбера я уже по-тихому начал работать. При знакомстве со мной В.С. Гостев сказал, что с нуклеопротеидами мы работать будем, а с Зильбером – никогда.
Но в эту пересменку сотрудники Зильбера свели меня со Львом Александровичем, и он в приватном порядке переговорил с В.Д. Тимаковым, тогда – директором Института, и тот перевел меня в отдел Зильбера, одновременно повысив в должности до старшего лаборанта. Это повышение резко подняло мой семейный жизненный уровень и существенно облегчило жизнь. Я сразу же включился в работу по фракционированию нуклеопротеидов, а Л.А. этому не препятствовал и вполне одобрял.
Сепараторы. Электронная микроскопия
Для фракционирования на РНК- и ДНК-нуклеопротеиды необходимо было вначале разделить тканевой гомогенат дифференциальным центрифугированием на ядерную фракцию, РНК- содержащие цитоплазматические гранулы (митохондрии и микросомы) и растворимый цитозоль. Для выделения гранул нужна была высокоскоростная центрифуга на 18.000 об./мин., что было тогда очень большой редкостью. Дифференциальное центрифугирование было введено несколько лет назад (1945–46 гг.) бельгийским биохимиком Альбертом Клодом (Claude), впоследствие Нобелевским лауреатом. Я познакомился с его работами еще в Университете и они, требующие высокоскоростной центрифуги, были предметом нереальных мечтаний. Но, когда я еще работал у Благовещенского, в его отделе была английская центрифуга MSE, универсальная, на средние обороты, очень удобная и хорошая. Как она попала в отдел, я не знаю – скорее всего, по ленд-лизу во время войны как медицинское оборудование.
В институт была направлена бригада инженеров НИИХимМаш'а, руководимая инженером Г.С. Безверхим, для копирования и воспроизведения центрифуги в СССР. Инженеры копировали MSE, а я поближе познакомился с Г.С. Он был человеком азартным, увлекающимся и рискованным. Был он специалистом по молочным сепараторам, но когда огляделся и увидел, что в микробиологическом институте есть проблема получения бактериальной массы, то установил тарелочные сепараторы (см. ниже) и с большим успехом стал применять их для осаждения бактерий и получения больших количеств бактериальной массы. Это было действительно замечательно и дирекция института, вернее, директор производства Н.Е. Лебедев, человек очень энергичный и влиятельный, вместе с Безверхим начали внедрять сепараторы в производство. Тогда же мы с Безверхим обнаружили в отделе скоростную насадку для MSE и после некоторых кустарных преобразований запустили ее в ход. Но я был уже у Зильбера, а MSE с насадкой – у Гостева. Правда, по-тихому, по вечерам, мы с Безверхим запускали насадку, а потом он приспособил ее к Зильберовской центрифуге с охлаждением, но работать на кустарном приспособлении было небезопасно, а, если бы что-либо случилось с центрифугой, мне бы не сносить головы. Притом Зильбер был особый любитель техники и очень бережно к ней всегда относился. Он свято верил в технику, а в начале 60-х считал, что, если бы у него была ультрацентрифуга «Spinco», то вирусная теория рака была бы быстро и полностью доказана (5).
Безверхий, энтузиаст сепараторов, предлагал мне попробовать осаждать на них наши частицы, и я попробовал. Стало ясно, что сепаратор может осадить из гомогената значительную часть фракции цитоплазматических гранул. При этом сепаратор работал с большими объемами гомогената. На этой основе можно было разрабатывать схему фракционирования опухолевой ткани на ядерную, цитоплазматические и растворимые фракции.
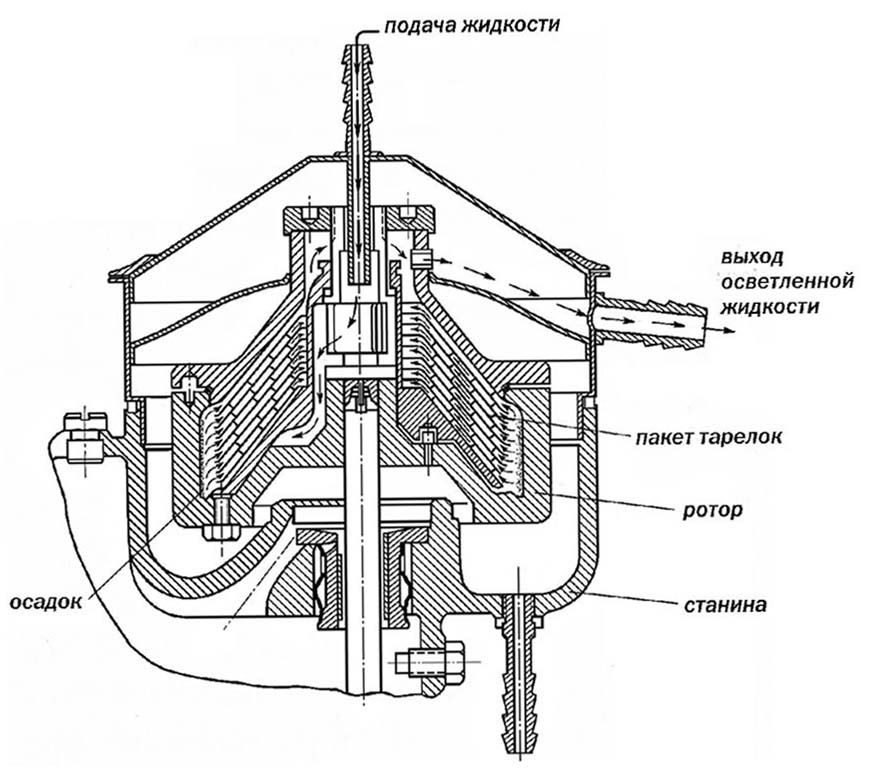
Рис. 1. Принципиальная схема тарелочного лабораторного сепаратора: разрез станины с ротором |
Принцип действия сепаратора основан на том, что сепарируемая суспензия поступает во вращающийся ротор сепаратора и распределяется там в пакете тарелок, отстоящих друг от друга с зазором ~ 1,5 мм [ Рис. 1 ]. Протекая между тарелками, жидкость образует тонкослойный ламинарный поток из которого идет осаждение частиц. Скорость вращения ротора (барабана) сепаратора достигает 20.000 об/мин., что дает высокоэффективное осаждение цитоплазматических гранул из большого объема (1 – 1,5 л) тканевого гомогената. Для лабораторных целей был сконструирован и изготовлен специальный сепаратор АСЛ-2 с небольшим барабаном из нержавеющей стали, высокоскоростной и с охлаждением. [ Рис. 2 ] .
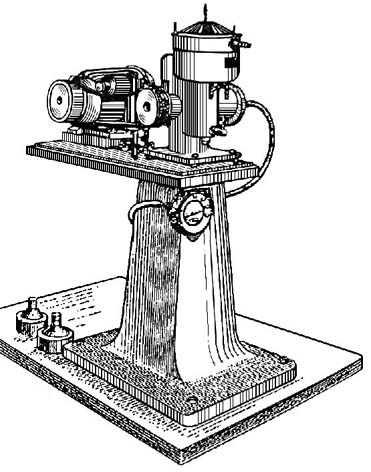
Рис. 2. Лабораторный сепаратор АСЛ-2. Общий вид. (Новикова и др., 1956 [11]) |
К сожалению, лабораторные сепараторы не были внедрены в серийное производство и их применение ограничилось в основном нашей лабораторией. В мировую лабораторную практику сепараторы не вошли, хотя интерес к ним и желание приобрести у наших и иностранных ученых было большим
Но параллельно я, под сильным давлением Л.А., вынужден был заняться поиском вирусов в опухолях с помощью электронного микроскопа. Тогда появился в институте первый электронный микроскоп, ЭМ-30, и наш институтский фотограф и микроскопист Николай Николаевич Соловьев освоил его, а Л.А. был полон желания и уверенности увидеть вирус в опухолях. Он очень настойчиво направлял меня на эту работу, и мне ничего не оставалось как заняться этими поисками параллельно с сепараторными делами.
Делалось это тогда следующим образом: препарат помещался на формваровую пленку, прозрачную для электронного пучка, и просматривался в вакууме на люминисцирующем экране. Поглощающие электронный луч структуры были на экране темными. Ясно, что препарат на пленке должен был быть свободен от солей и любых растворимых компонентов, которые при высыхании неизбежно создавали бы непрозрачный осадок. Для освобождения от солей и даже растворимых белков применялся «капельный диализ» – остроумный метод, в котором капля, содержащая исследуемый объект, наносилась на формваровую пленку, формируемую на поверхности дистиллированной воды. При этом за 2-3 часа проходил диализ, освобождающий препарат от солей и частично от растворимых белков. Очевидно, что диализ приводил к осмотическому шоку клеток, например, к гемолизу эритроцитов и возникновению ряда артефактов. Я применил принцип «ультрафильтрации» [ Рис. 3 ], при котором формваровая пленка формировалась на изотоническом растворе NaCl, содержащем исследуемый объект, и помещалась затем на ацетат-целлюлозную мембрану. Солевой раствор всасывался через пленку в мембрану, а объект оставался на пленке без контакта с дистиллированной водой. Таким образом, эритроциты идеально сохраняли свою нативную форму, а лизирующиеся микрококки можно было наблюдать на различных стадиях лизиса (Абелев и Соловьев, 1953). Это была моя первая журнальная публикация.
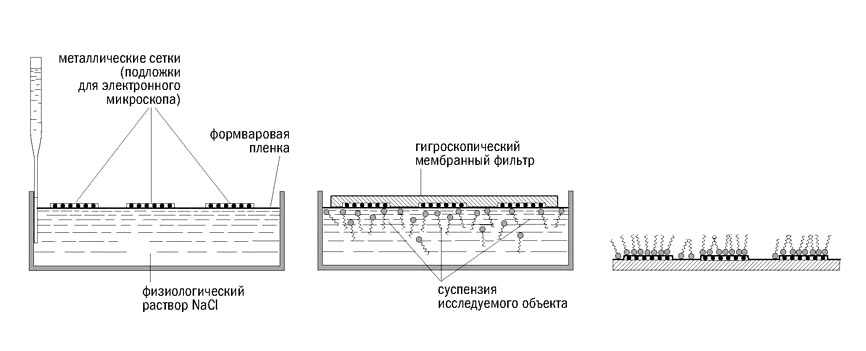
Рис. 3. Приготовление препарата (Объяснение в тексте) |
Для того же, чтобы увидеть вирус, надо было получить в ультрацентрифуге его концентрированный и очищенный препарат. Но ультрацентрифуги у нас не было, и мы воспользовались методом осаждения вируса метанолом. Из опухоли, вызванной у кролика вирусом папилломы Шоупа, мы получали метаноловый преципитат, который содержал частицы, очень сходные с частицами этого вируса, описанными в литературе. Л.А. поместил электронную фотографию вирусных частиц в одном из своих популярных обзоров в «Природе» (Зильбер, 1955). Дальнейшая работа по метаноловым преципитатам проводилась В.А. Парнес на лейкозах человека (В.А. Парнес, 1956, 1960) (6). Я же от этой работы отошел – у меня не было ни чувства твердости, ни надежности в этом подходе и результатах.
В это время у Л.А. было убеждение, что если хорошо посмотреть, то под электронным микроскопом вирусы в опухоли будут просто видны. Но еще не было ультратонких срезов и, чтобы увидеть вирусы, надо было их сначала выделить и очистить под контролем биологических тестов. Пока же было время «вирусоподобных частиц».
Несколько позже (1955 г.) когда в отделе появился свой ЭМ-30, Л.А. посадил на эту работу А.И. Гусева. Толя долго мучился, воспроизводил результаты А.А. Шубина из ИЭПИТР'а (7), на чем настаивал Л.А., пока не убедился, что вирусоподобные частицы из опухолей, сходные с описанными А.А. Шубиным, – это частицы стекла, применявшиеся для растирания тканей (Гусев, 1959). Таким образом, «стеклянный вирус» закончил эпопею «вирусоподобных частиц» в опухолях, и у Л.А. пропал интерес к этой проблеме.
Тем временем работы с сепаратором продолжались и давали все возрастающую уверенность в его применимости для выделения цитоплазматических гранул. На этой основе была разработана схема фракционирования гомогената, сочетающая получение ядерной фракции низкоскоростным центрифугированием, затем фракции митохондрий и микросом – на сепараторе, и разделение растворимых белков последовательным высаливанием сернокислым аммонием. Схема была довольно полная, даже красивая, хотя и требовала еще серьезных доработок. Одна из них – вспенивание белкового раствора при выходе его на большой скорости из барабана сепаратора. Это вспенивание вело к денатурации белков и образованию уже артефактного осадка, вероятно, искажающего результаты фракционирования. Но так или иначе схема стала основой моей работы: полученные фракции я анализировал электрофоретически в аппарате Тизелиуса (8) и передавал Николаю Васильевичу Нарциссову для проверки на реактивность с сыворотками крыс – носителей первично-индуцированных или перевиваемых опухолей. Эта серия работ легла в основу моей кандидатской диссертации (Абелев 1954), а статья по первому сепаратору была опубликована в сборнике, посвященном 60-летию Л.А.Зильбера (Абелев и Безверхий, 1956). Надо сказать, что вся эта работа протекала в разгар «Дела врачей», борьбы с «космополитизмом», нависшего увольнения и тяжелого материального и семейного положения. Но это уже другой сюжет, а защита диссертации, перевод в тот же день в младшие научные сотрудники со степенью и начавшаяся еще дохрущевская «оттепель» резко изменили жизненную и рабочую ситуацию. Конечно, в моей работе того периода было много кустарного и неясного, но применение сепараторов для фракционирования тканевых гомогенатов безусловно было «сухим остатком». Г.С. Безверхий был полон энергии и энтузиазма, он привел двух инженеров (Е.С. Новикову и В.М. Джинчарадзе) и слесаря-универсала Павла Гарнова специально для разработки лабораторного сепаратора для наших целей, о чем я уже писал выше, и вскоре такой сепаратор (АСЛ-2) – небольшой по размеру и объему обрабатываемого материала, скоростной (до 20.000 об./мин.), из нержавеющей стали и с охлаждением – был сделан [ Рис. 2 ] и сразу же вошел в работу (Новикова и др., 1956 а). Мы долго им пользовались. Инженеры глубоко прониклись нашими задачами. Вскоре возникла идея применить для фракционирования гомогенатов камерный сепаратор, принцип которого, кажется, был уже известен. Идея этой машины заключалась в том, что поток жидкости, содержащей осаждаемые частицы, последовательно протекал «змейкой» через цилиндры увеличивающегося радиуса, расположенные во вращающемся барабане сепаратора. Осаждение велось из тонкого ламинарного слоя жидкости, протекающего мимо стенок цилиндров, имеющих увеличивающийся диаметр, т.е. возрастающую центробежную силу [ Рис. 4 ].
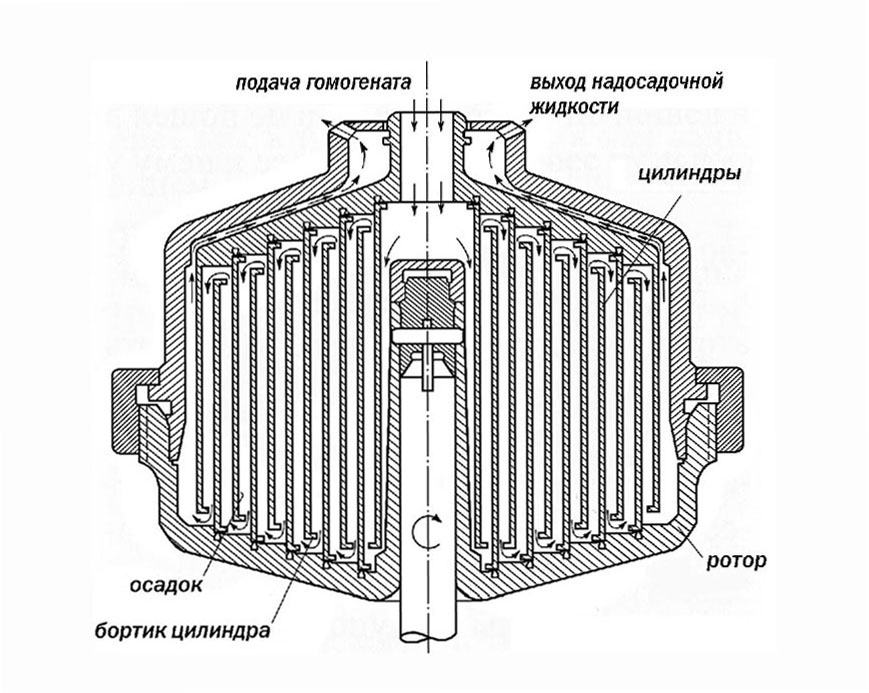
Рис. 4. Схема ротора камерного сепаратора |
Таким образом, камерный сепаратор сочетал высокую эффективность осаждения с фракционированием частиц по скорости осаждения, т.е. по размеру. Принципиально важным усовершенствованием метода, введенным в нашей работе, была установка растворов сахарозы различной плотности между потоком жидкости и стенками цилиндров. Это создавало возможность одновременного разделения частиц по размеру и по плотности. Мы придумали простой метод определения плотности слоев сахарозы во вращающемся барабане и способы установки слоев различной плотности в разных цилиндрах. Камерный сепаратор позволял в одном цикле одновременно получать несколько фракций из гомогената печени без выхода из вращающегося барабана, т.е. без вспенивания. Мы привлекли А.И. Гусева для электронно-микроскопического анализа получаемых фракций и вскоре имели схему одновременного разделения гомогената печени кролика на ферритиновую, митохондриальную, гликогеновую и микросомную фракции (Новикова и др., 1956 б). Наш сепаратор не имел аналогов в очень обширной в то время литературе по разделению цитоплазматических гранул центрифугированием (см. Абелев, 1962) (9). Был снят фильм об этом методе, и мы демонстрировали его на Всесоюзном Съезде Микробиологов в 1956 г. и на Х сессии АМН СССР (Абелев и др. 1959). К сожалению, тогда не было лабораторной промышленности, и сепаратор не пошел в серийное производство, несмотря на серьезный интерес к нему у нас и за границей.
Камерный сепаратор был отличным финалом этого цикла работ, но мы уже переходили на иммунодиффузионный анализ, а среди инженеров возникли сложные отношения и раздоры. Смерть одного из министерских начальников, заинтересованного в этой работе (Джинчарадзе), а потом и директора завода «Физприбор» Д.В. Укладова, большого поклонника Л.А. Зильбера и его исследований, сильно затормозили эти работы, которые вскоре и совсем прекратились.
Но мы уже были полностью поглощены иммунодиффузией, в которой прежде всего использовали фракции, получаемые на сепараторе.
Иммунодиффузия: индивидуальные антигены
Предистория этого цикла работ была такова. В 1947 г. Л.А. разработал и применил метод анафилаксии с десенсибилизацией (А-Д) для обнаружения в опухолях специфических для них антигенов. Эти антигены были обнаружены в ходе поисков опухолеродных вирусов. Поэтому исследовались нуклеопротеидные фракции опухолей. В методе А-Д морские свинки сенсибилизировались нуклеопротеидом из опухоли, затем десенсибилизировались аналогичной фракцией нормальной ткани, а в качестве разрешающего введения вводился исходный препарат, вызывая, как правило, сильную анафилактическую реакцию. Казалось, что нужно только определить, какая фракция разрешающего препарата будет ответственна за реакцию, и природа опухолевой специфичности будет определена. Это и представлялось мне основной задачей, и первым делом, как говорилось выше, необходимо было разделить и определить активность РНК- и ДНК-нуклеопротеидов. Задача оказалась практически неразрешимой. Здесь не место анализировать возможные причины (10), но все попытки оказывались как в трясине: сколько активных фракций было в разрешающем препарате? Каковы количественные отношения между десенсибилизирующим и разрешающим введением? Какова роль индивидуальности животных в ответе на отдельные антигены? И много других вопросов. Главное, что не было ни твердости, ни воспроизводимости результатов при попытках определить природу антигена(ов), ответственного(ых) за А-Д. Метод демонстрировал антигенную специфичность опухолей, но едва ли был пригоден для биохимического анализа феномена.
Изучать антигены, реагирующие в реакции связывания комплемента (РСК) с сыворотками крыс – носителей опухолей, было и проще и надежней. Эту систему разрабатывал Николай Васильевич Нарциссов, старый сотрудник Л.А., серолог, необычайно тщательный и скрупулезный исследователь. Наша работа пошла именно по этому пути (Зильбер и др., 1955; Нарциссов и Абелев, 1956 а, б, в). Но и этот путь не был достаточно ясным и информативным. Даже в том, что граница между специфической реакцией и так называемой антикомплементарностью (11) антигенов была весьма зыбкой и сильно зависела от концентрации антигена и свойств сыворотки. Мы пытались обставить анализ разными контролями, но постоянно оставалось ощущение «хождения по краю». На этом фоне произошло знакомство с преципитацией в геле, вернее с его главным вариантом – двойной диффузией в геле, только что предложенным Ухтерлони (Ouchterlony). Первыми работами по этому методу и его применению к опухолям были две работы В.Björklund & V.Björklund (1952) (12), появившимися в печати еще до основной публикации Ouchterlony (1953) (13). Насколько я помню, о них на конференции лаборатории сообщила В.А. Парнес, как о некой новой методике. Проверить ее было предложено Зине Чирковой, ст.лаборанту биохимической группы, девушке весьма пассивной и пугливой. У нее, конечно, ничего не пошло. Помимо нового принципа, в этом методе для биохимика все было ново – сильные преципитирующие сыворотки, агаровый гель и сам принцип двойной диффузии. Не помню как, – может быть, помогая Чирковой разобраться в методе, – я вдруг понял всю красоту и потенциальные возможности метода и загорелся желанием попробовать его.
Метод был основан на чрезвычайно простом принципе – диффузии индивидуальных антигенов и антител, помещенных в геле агара напротив друг друга. При встрече в геле каждая пара антиген-антитело давала свою полосу преципитации. Таким образом, при диффузии смеси антигенов навстречу смеси антител, число полос соответствовало числу пар антиген-антитело. Если против лунки с антителами под углом друг к другу помещались лунки сравниваемых антигенов, то в случае их идентичности полосы преципитации образовывали сливающуюся дугу. В случае неидентичности они пересекались, а при частичном родстве образовывали «шпору» со стороны гомологичного антигена.
Таким образом метод позволял определять антигенный состав сложных природных смесей (крови, тканевых экстрактов) и сравнивать друг с другом по специфичности антигены, не выделяя их из смеси.
Главная трудность была в преципитирующей сыворотке, получение которой казалось тогда для биохимика задачей, далеко выходящей за пределы его компетенции. Причем, не только казалось, но и действительно было.
В отделе тогда была такая сыворотка, против лейкемической селезенки человека – у З.Л. Байдаковой, опытнейшего сотрудника-иммунолога, правой руки Л.А. Этой сывороткой лизировали лейкемические клетки для приготовления соответствующей вакцины. Лейкозами тогда занималась В.А. Парнес, которая была специалистом по лейкозам. Мы быстро договорились с В.А. и начали воспроизводить работу Бйорклундов на лейкозах человека. Главное было увидеть полосу преципитации, и мы ее увидели! Она была одна (или две?), но была, – метод пошел! Мало того, что полоса была – она давала реакцию идентичности. Сразу же, конечно, стало ясно, что реакция малочувствительна – один-два антигена в экстракте лейкемической селезенки были, конечно, сáмой вершиной айсберга, и казалось, что, вряд ли, эта реакция окажется способной обнаружить специфические опухолевые антигены. Но мне стало ясно, что реакция бесценна в сравнении состава внутриклеточных фракций – ядер, митохондрий и микросом, и здесь вопросы ее чувствительности имели не самое главное значение. Любые специфичные или общие для этих фракций антигены были крайне интересны, и ничего подобного биохимия того времени дать не могла. Я же как раз и сравнивал тогда с помощью электрофореза белки ядер, митохондрий, микросом и цитозоля, и имел по этому поводу некоторые представления об их возможном родстве (Абелев, 1954, 1956).
Мы договорились с В.А., что все, что касается сравнения опухолевой и нормальной ткани – область ее интересов, а сравнение антигенной структуры внутриклеточных фракций – моих. Мы приступили к этой работе, а вскоре вместе с З.А. Авенировой и З.Л. Байдаковой начали готовиться к работе на мышиной печени – классическом объекте изучения внутриклеточных органелл и хорошо уже мне знакомому по работе с сепараторами. Но Парнес резко воспрепятствовала этой работе, разразился скандал, очень для меня тяжелый и болезненный, и я с трудом вырвался из этого «союза», оставив ей все материалы по лейкозам, полученные в совместной работе.
Придя в себя, мы энергично включились в иммунодиффузионный анализ печеночных фракций, одновременно вникая и усовершенствуя саму иммунодиффузию. Мы стремились увеличить спектр полос, подвластных анализу, наладить фоторегистрацию результатов, оптимизировать условия сравнения антигенов (спектров полос преципитации), понять, от чего зависит чувствительность метода и как можно ее увеличить. Начали налаживать микрометод. Мы упивались иммунодиффузией и ее потенциальными возможностями, что совпадало с повсеместным распространением метода и его усовершенствованием. Первая успешная работа по иммунодиффузионному анализу печеночных митохондрий и микросом привела к обнаружению органоспецифического антигена печени и его утраты в перевиваемой гепатоме (Абелев и сотр., 1959). Одновременно с антигенным упрощением был получен, скорее, намек на обнаружение специфического антигена гепатомы (Зильбер и сотр., 1959). Очень актуальным стала оптимизация сравнительного анализа полных антигенных систем, т.е. систем антиген-антитело. Простым и эффективным решением стала постановка в «четверках» (Абелев, 1960), позволившая сравнивать две полных системы антиген-антитело и «переместить» наиболее важный для сравнения район реакции – район образования «шпор» – с периферии в центр, где полосы видны наиболее четко. Более того, линии, образованные антигенами, общими для сравниваемых систем, шли под 45° к линиям, уникальным для одной из систем, а эти последние образовывали прямой угол друг с другом [ Рис. 5 ].
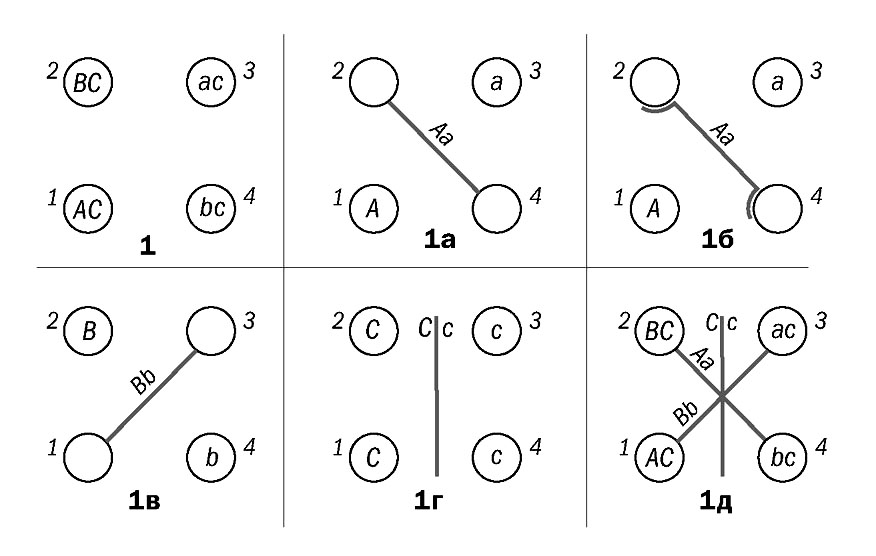
Рис. 5. Схематическое изображение постановки «по квадрату»: А, В, С - иммунологически неродственные антигены; а, b и с - антитела к А, В и С соответственно; Аа, Вb, Сс - линии преципитации в геле. На схеме 1б антитело а - в избытке по отношению к антигену А (Абелев, 1960) |
Постановка в «четверках» была опубликована и за границей (Abelev, 1960), она очень быстро распространилась, вошла в набор штампов (матриц) для постановки реакции, и с легкой руки англичанина Файнберга (J. Feinberg), энтузиаста иммунодиффузии и автора набора штампов для иммунодиффузии, получила название «squaire pattern» – постановки по «квадратной схеме». В 1959 г. Файнберг посетил отдел Л.А., я рассказывал ему о «четверках», что ему очень понравилось. Он пригласил меня в Лондон на Ciba Foundation Symposium по преципитации в геле (1959 г.). Меня, естественно, не пустили, а Файнберг рассказал на симпозиуме о четверках, что, как он мне написал, вызвало интерес у участников. «Четверками» я горжусь наравне с сепараторами.
Важным вкладом в иммунодиффузию (ИД) была работа по технике ее микрометода, доведенная до совершенства Гусевым и Цветковым (1961).
Работа с иммунодиффузией началась в 1956 г., а в 1958-1959 гг. она вышла на экспоненту как в результативном, так и в методическом плане. На результатах я остановлюсь в других разделах, а здесь расскажу о прогрессе в методической области. Ясно, что наряду с иммунопреципитацией мы думали и об иммуноэлектрофорезе (ИЭФ). В 1959 г. Петр Николаевич Грабар, иммунохимик из Института Пастера в Париже, автор ИЭФ приезжал в Москву и читал лекции об ИЭФ. Мы уже много работали со свободным электрофорезом, а в 1957 г. я был в Праге, работал там вместе с аспирантом института Органической химии Зденеком Прусиком по препаративному электрофорезу в свободной среде, но попытки наладить ИЭФ нам не удавались. ИЭФ ставили в пластинке агарового геля, залитого на стекле и соединенного мостиками из фильтровальной бумаги с электродными сосудами, содержащими буферный раствор. Гель на катодной границе с переходником из фильтровальной бумаги пересыхал и электрофорез прекращался. Стало ясно, хотя и не сразу, что причиной этого является различие электроосмотического потока на границе двух пористых сред, и мы изменили конструкцию прибора так, что бумажные переходники были исключены, а пластинка геля контактировала с буферными растворами через гелевые отсеки (Абелев и Цветков, 1960) [ Рис. 6 ].
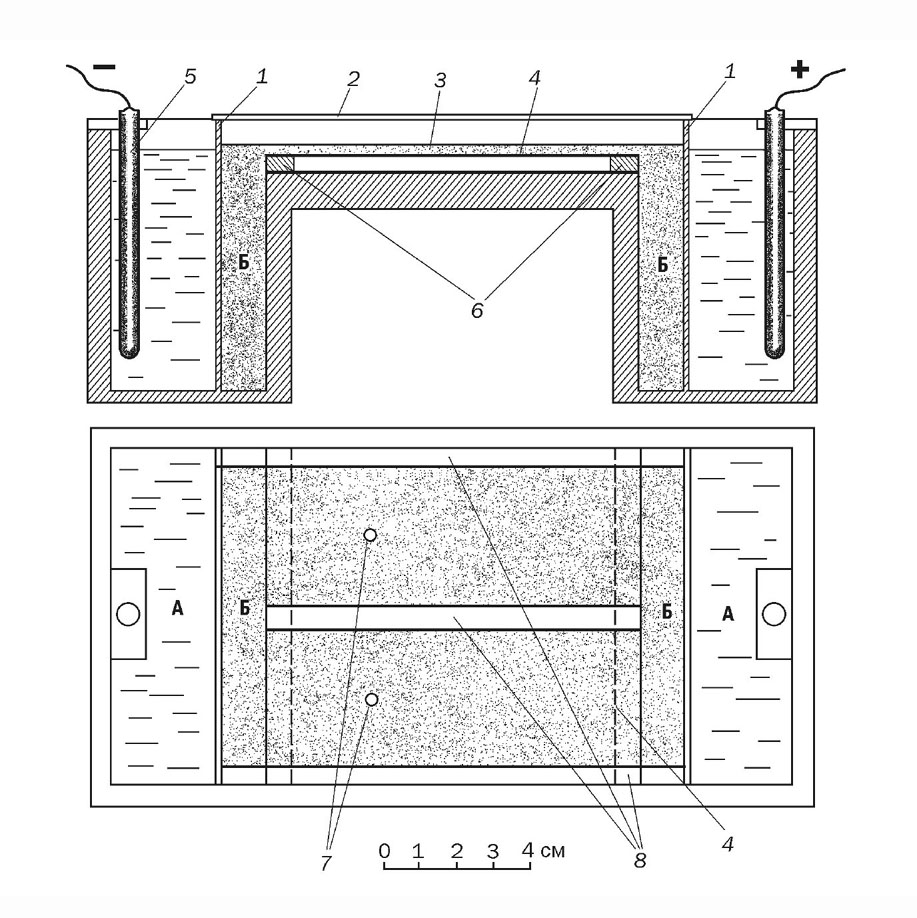
Рис. 6. Схема аппарата для аналитического иммуноэлектрофореза. А – секция электродного сосуда, заполненная буфером; В – секция электродного сосуда, заполненная агаром; 1 – рамка с шелковым ситом; 2 – крышка; 3 – агаровая пластинка на стекле для электрофореза; 4 – стеклянная подложка; 5 – электрод; 6 – бортики для стекла; 7 – отверстия в агаре для внесения препарата; 8 – стеклянные бортики на стекле для электрофореза. (Абелев, Цветков, 1960) |
Соответствующий прибор сделал нам наш неизменный помощник, холодильный мастер Николай Сергеевич Федотов («Федотыч»), а в лаборатории с ИЭФ мы стали работать вместе с В.С. Цветковым, также весьма технически искушенным сотрудником. ИЭФ пошел и стал элементом повседневной работы (Цветков, 1965; 1968).
Очень важным этапом стал метод получения моноспецифических антител («элюатов») к индивидуальным антигенам. Применив едва ли не самый простой и самый первый в иммунохимии способ извлечения антител из преципитатов в кислой среде, мы получили антитела из преципитата органоспецифического антигена печени (АО), а затем, пользуясь истощенными моноспецифическими антисыворотками, – из различных органоспецифических и опухоле- специфических преципитатов (Абелев и Авенирова, 1960). Если специфический преципитат нельзя было получить из «истощенных» гетерологическими антигенами сывороток, содержащими антитела к нескольким антигенам, то экстракт вначале разделяли препаративным электрофорезом в агаровом геле так, чтобы можно было составить пары антиген-антитело, направленные к одному антигену (Khramkova and Abelev, 1963). Таким способом, нами была получена коллекция моноспецифических антител к антигенам печени мышей, ставшая основой соответствующего исследования судьбы этих антигенов в гепатомах мышей (см. раздел об органоспецифических антигенах). А.И.Гусев с успехом использовал этот метод для получения элюата антител к специфическому клеточному антигену саркомы Рауса (Гусев, 1960 б).
Это был критический этап работы. Мы перешли на исследование индивидуальных антигенов, не выделяя их из сложной, вернее сложнейшей системы тканевых экстрактов или внутриклеточных органелл. Тест-система состояла из элюата антител или моноспецифической сыворотки к определенному антигену и тканевого экстракта, взятых в оптимальном разведении (Храмкова и Абелев, 1961). При подстановке к тест-системе исследуемого препарата можно было точно идентифицировать в нем исследуемый антиген и по его титру определить полуколичественно содержание антигена, сравнительно с его концентрацией в тест-системе. Но здесь возник очень существенный вопрос о чувствительности тест-системы. Чувствительность любой тест-системы, т.е. полосы преципитата, ею образуемый, составлял 1/8 – 1/16 от концентрации антигена или антитела тест-системы. Более низкие уровни этих реагентов не отклоняли полосу тест-системы, т.е. не обнаруживались ею. Это относительный предел чувствительности тест-системы. Если же ее разбавлять, то относительный предел чувствительности будет сохраняться, выявляя все более низкие концентрации антигена (или антител). Предельное разведение тест-системы, дающее еще видимую полосу преципитации будет обладать наибольшей чувствительностью в определении абсолютной концентрации антигена – и это будет абсолютный предел. Мы сначала «вычислили», что чувствительность реакции будет тем выше, чем слабее тест-система, а потом очень убедительно показали это экспериментально (Храмкова и Абелев, 1961; Куприна, 1965), одновременно увеличив абсолютный предел чувствительности обработкой преципитатов солями Cd++. Абсолютный предел чувствительности для антигенов с мол. весом IgG был ~ 1 мкг/мл, а для более «тяжелых» 3–5 мкг/мл. Тест-системы стали основой нашего анализа.
Серьезным прорывом и важным элементом иммунохимического анализа стал метод иммунофильтрации антигенов. К этому методу мы пришли в процессе выделения специфического антигена гепатомы (АГ). На след АГ мы «напали» в первых же работах по сравнению антигенов печени и гепатомы мышей с применением «четверок» (Зильбер и сотр., 1959). Это была слабая «тень» специфического антигена, но Л.А. интересовался именно ею. Мы видели полосу антигена гепатомы (АГ) в препарате цитоплазматических гранул, получаемых в сепараторе, но как-то, когда мы с З.А. Авенировой уехали в Ленинград на конференцию, Наташа Энгельгардт, недавно вошедшая в нашу группу, оставшись одна, поставила вместо препарата из гранул, просто экстракт гепатомы и получила настолько яркую картину прецитипации АГ, что мы сначала этому результату не поверили, а потом, подтвердив его, полностью перешли на экстракты. Сепараторы остались только для получения одного (правда, самого четкого и сильного) органоспецифического антигена печени (Абелев и сотр. 1960). Так логика нашей работы увела нас от сепараторов. Мы стали выделять АГ из экстракта высаливанием, но он шел с большими примесями печеночных и сывороточных антигенов, и тут возникла мысль применить антитела к примесям, т.е. к антигенам печени, для очистки АГ от этих примесей. АГ оказался антигеном с высокой электрофоретической подвижностью, он шел непосредственно за сывороточным альбумином. Стало ясно, что, если поместить антитела к нормальной печени при электрофорезе перед АГ, то можно «профильтровать» его через антитела и преципитировать или сильно замедлить миграцию примесей, очистив таким образом АГ. Мы уже хорошо знали тогда, что иммуноглобулины переносятся в агаровом геле электроосмосом к катоду и позволяют тем самым создать встречное движение АГ и антител в геле, т.е. обеспечить иммунофильтрацию. Вместе с В.С. Цветковым мы сразу же стали разрабатывать препаративный вариант метода. Но, поместив на пути АГ иммуноглобулиновую фракцию анти-печеночной сыворотки, мы почти наверняка загрязнили бы отфильтрованный АГ «быстрыми» примесями, содержавшимися в «фильтре», и вместо одних примесей получили бы другие. Для устранения этого, а заодно, чтобы избежать получения иммуноглобулинов из антисыворотки «фильтра», мы вначале подвергали электрофорезу цельную антисыворотку и включали иммуноглобулин в катодную часть геля, после чего вырезали всю его анодную часть и заливали свежим гелем. Траншею АГ делали в свежем геле, затем полюса переключали, и все, что вошло в гель благодаря электроосмосу, возвращалось в исходное положение, в то время как «профильтрованный» АГ оказывался в зоне геля, свободной от примесей и от белков фильтра. Метод работал, но мешало еще одно, казалось бы, непринципиальное препятствие. Фракция, подлежащая фильтрации, заливалась в смеси с агаром в траншею в геле. При электрофорезе траншея лопалась или трескалась, и разделение искажалось. Эфект был непостоянным, но он делал метод неаккуратным и плохо воспроизводимым. Мы перепробовали разные способы зарядки и «реставрации» траншей во время электрофореза, но ничего не помогало, пока не осенила простая мысль – все дело в различии скоростей электроосмотического потока в геле и в траншее с антигеном, и это различие искажает гель. А поток определяется напряженностью поля, которая в свою очередь, – сопротивлением, а оно – концентрацией соли, отличной от окружающего геля. Немедленная проверка полностью подтвердила и воспроизвела картину артефакта, а тщательный диализ вносимого материала полностью устранил артефакт. Метод был готов. Он дал четкие и хорошие результаты. Очищенный АГ был в руках (Абелев и Цветков, 1960; Abelev, Zvetkov, 1961; Зильбер и Абелев, 1962).
Было очевидно, что метод был универсальным, но не мог быть применен к «медленным» антигенам с подвижностью γ-глобулина. Модификация γ-глобулина «фильтра» изотиоцианатом или арсаниловой к-той, не влияя на способность антител связываться с антигеном, меняла существенно их подвижность (создавала анодную подвижность в агаре) и тем самым обеспечивала их «фильтрующую» способность. Этим способом удавалось, например, разделить мышиный и человеческий IgG, имеющие почти равные подвижности и не отличающиеся сколько- нибудь существенно от IgG фильтра (см. Зильбер и Абелев, 1962; Цветков, 1964, 1965) (14).
При разработке этого варианта метода нами самым широким образом применялся аналитический вариант иммунофильтрации [ Рис. 7 ], получивший в дальнейшем самостоятельное развитие в том числе и для выявления зоны изучаемого антигена непосредственно в геле (Абелев, 1965, 1966).
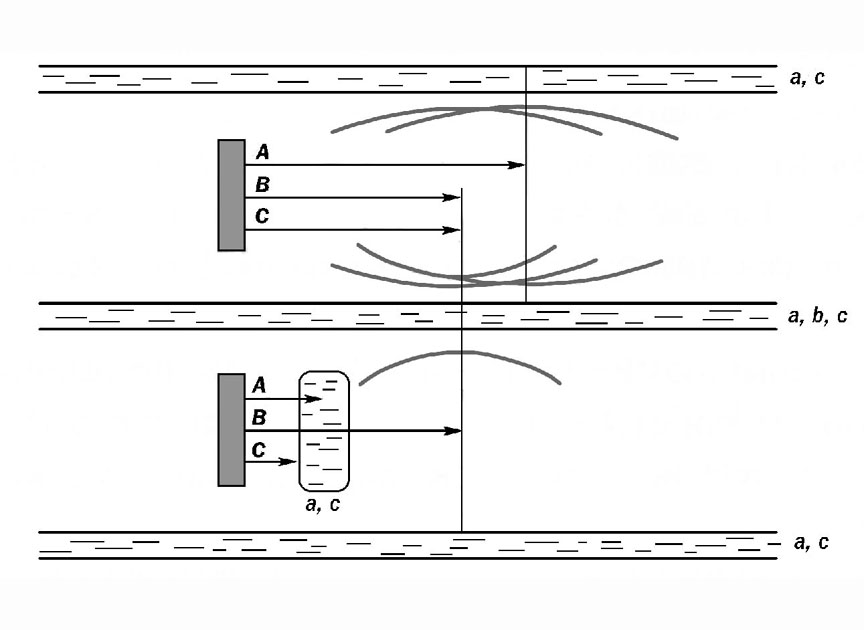
Рис. 7. Принцип метода аналитической иммунофильтрации. А, В, С - антигены, не дающие перекрестных реакций друг с другом; a, b, c - антитела к этим антигенам. А и С блокируются антителами «фильтра», антиген В беспрепятственно проходит сквозь «фильтр». После иммунофильтрации в агаре выявляется лишь антиген В. В контроле образуются дуги всех антигенов, содержащихся в препарате. (Абелев, 1965) |
Итак, в виде стройной схемы иммунодиффузионная система анализа антигенной структуры тканей «на уровне индивидуальных антигенов» была создана к началу 60-х годов и применялась уже не только для печени и гепатом, но для саркомы Рауса и канцерогенных опухолей кур и, отчасти, для рака желудка человека (Цветков и Людоговская, 1964). В систематической форме она была представлена в гл. 14, 15 и 18 монографии «Вирусология и имммунология рака» Л.А. Зильбера и Г.И. Абелева (1962) и в ее дополненном английском варианте (Zilber & Abelev, 1968), а также в развернутом автореферате моей докторской диссертации (Абелев, 1963). «Четверки» широко применялись в иммунохимическом анализе в исследовании самых различных антигенов, аналитическая иммунофильтрация была включена в виде отдельной главы в монографию E. Day, Cancer Immunochemistry, 1965 (см. Абелев, 1966). Подробное описание почти всех наших методов вышло позже в руководстве «Иммунохимический анализ» п/р Л.А. Зильбера (1968), служившим настольной книгой по иммунохимии едва ли не до 90-х годов. Иммунодиффузия, с ее теоретическим обоснованием, анализом и многими модификациями, равно как с практическим выполнением – начиная с приготовления агара и до регистрации результатов – вошла в иммунохимическую практику у нас в стране, главным образом благодаря опыту лаборатории и через него. Этому немало способствовали ежегодные семинары, проводимые в лаборатории, постоянно идущие стажировки и практикум по курсу иммунохимии на кафедре вирусологии МГУ, организованный Н.А. Дорфманом в 1965 г. и постоянно действующий до настоящего времени (2005 г.), естественно, с изменениями и дополнениями.
Иммунодиффузия со всеми ее вариантами обеспечивала иммунохимический анализ на уровне индивидуальных антигенов. Но для полноты картины он должен был сочетаться с локализацией индивидуального антигена в тканях, в определенных клеточных типах. Таким образом, очень важной частью системы иммунохимического анализа стала иммунолокализация антигенов на срезах с помощью моноспецифических антител. Этот метод применительно к тканевым антигенам не был еще освоен в стране. В пятидесятых – начале шестидесятых годов уже была налажена в нашем институте метка антител изотиоцианатом флуоресцеина, были флуоресцентные насадки к обычному микроскопу МБИ, а затем и очень хороший люминисцентный МЛ-2, но не было ни криостатов, ни опыта работы с иммунофлуоресценцией (ИФ) на срезах, равно как и с истощенными антисыворотками к тканевым антигенам.
По ходу исследований их логика привела нас к необходимости установить происхождение, т.е. исходную тканевую локализацию, как органоспецифических антигенов печени так и АГ. Вопрос тогда стоял так: является ли АГ антигеном эпителия желчных ходов или самих гепатоцитов? И на этот вопрос могла ответить только ИФ. А от ответа на него зависела судьба всего направления наших работ. В 1959–1960 гг. освоением и налаживанием ИФ занялась недавно вошедшая в нашу группу после окончания Университета ст.лаборант Н.В. Энгельгардт. С криостатом познакомились в лаборатории В.В. Португалова, нашего первого гистохимика, а собственный криостат, вполне пригодный для профессиональной работы, сделал для нас Н.С. Федотов, механик-холодильщик, замечательный человек и мастер, глубоко проникнувшийся нашими делами. Я уже писал о нем в связи с освоением иммуноэлектрофореза.
Первые работы по ИФ Н.В. Энгельгардт начала сразу с локализации АГ. Она воспользовалась антигепатомной сывороткой, абсорбированной экстрактом печени и выявлявшей одну полосу преципитации с экстрактом гепатомы, т.е. моноспецифичной по критериям иммунодиффузии. Результаты получились абсолютно неожиданные – на срезах ярко окрашивался внеклеточный матрикс, соединительная ткань (строма опухолей) и ничего более. Откуда? Почему? Вскоре мы поняли причину – антисыворотка абсорбировалась экстрактом (т.е. растворимыми антигенами) печени, испытывалась в иммунодиффузии с экстрактом (т.е. опять с растворимыми антигенами), а получена была к фракции цитоплазматических гранул, содержащих фрагменты нерастворимых структур стромы, эпителия и соединительной ткани. Антитела к ним не убирались при нейтрализации растворимыми антигенами, и их то мы в первую очередь и увидели (Энгельгардт и Абелев, 1962). Это предположение подтвердилось получением из таких сывороток элюата антител на нерастворимом осадке из гепатомы, дающем идентичную ИФ картину на срезах. Стало ясно, что абсорбированные антисыворотки, полученные иммунизацией тканевыми гомогенатами или их фракциями, не пригодны для ИФ или, по крайней мере, опасны в иммуногистохимических исследованиях. Здесь были необходимы элюаты антител, и первые же опыты с элюатами к четырем органоспецифическим антигенам печени дали вполне однозначные результаты – эти антигены локализовались только в гепатоцитах (Энгельгардт, 1963; Engelgardt et. al, 1963). При этом необходимым отрицательным контролем в ИФ было полное подавление свечения, при нейтрализации элюата антигеном в зоне эквивалентности (Энгельгардт, 1968). Но с элюатом к АГ ничего не получалось – он на срезах не определялся. В дальнейшнй работе, когда выяснилось, что АГ – белок сыворотки, синтезируемый в клетках и легко из них вымываемый, стало ясно что для него требуются особые условия фиксации, как например, для сывороточного альбумина. Когда эти условия были соблюдены, впервые АГ, который был уже тогда альфа- фетопротеином (АФП), был локализован в эмбриональной печени мышей и человека (Энгельгардт и др. 1969). Но дальше возникли более специфичные для АФП трудности. Он накапливался в сыворотке больного или животного с опухолью, и его концентрация там зачастую превышала концентрацию в опухоли. Он мог легко попасть в мертвые или погибающие клетки с нарушенной проницаемостью и быть там обнаружен. Как отличить синтез от пассивного захвата на срезах опухолей? Этот вопрос с особой остротой возник после работы Н.В. Энгельгардт и А.И. Гусева в Африке с гепатоцеллюлярной карциномой человека. Тогда и был предложен «γ-глобулиновый контроль», в котором серийные срезы окрашивались на АФП и эндогенный IgG человека, заведомо гепатомой не синтезируемый. Клетки с нарушенной проницаемостью содержали оба белка, выявляемые на двух последовательных срезах, в то время как живые клетки, продуцирующие АФП, содержали только его (Engelhardt et al., 1971). Таким образом, мы смогли отличить клетки, захватившие АФП, от синтезирующих его in vivo. Контроль на эндогенный IgG вместе с нейтрализацией антител в зоне эквивалентности стали необходимыми критериями в работе с иммунолокализацией АФП (Abelev et al., 1979).
Замечательным продолжением в изучении локализации антигенов явилась иммуноэлектронная микроскопия (ИЭМ). Она была начата в лаборатории Н.А.Дорфманом для обнаружения мембранных антигенов лейкемии мышей (Дорфман, 1971). В этом методе, требующем большого искусства, подлинных высот достиг В.Н. Баранов в изучении внутриклеточных антигенов (Baranov & Engelhardt, 1987). ИЭМ позволяет с точностью идентифицировать клетки, где локализован антиген, и клеточные структуры, с которыми он связан. Он позволяет судить о том, синтезируется ли антиген в данных клетках, или попадает в них путем рецептор-опосредованного эндоцитоза, или пиноцитозом, или неспецифической диффузией (см. «АФП – биология»). В наших работах ИЭМ оказалась бесценной в изучении АФП в регенерирующей печени и характеристике мышиного аналога одного из членов семейства раково-эмбрионального антигена человека (BgP) (15) (Kuprina et al., 1990). Работы В.Н. Баранова относятся к лучшим международным исследованиям с использованием ИЭМ.
Таким образом, ИФ и иммуноэлектронная микроскопия (вернее, иммуногистохимия в ее различных вариантах) вошли в систему иммунохимического анализа на уровне индивидуальных антигенов как ее необходимый и завершающий этап.
Специфической задачей в изучении индивидуальных антигенов было исследование их биосинтеза in vivo и in vitro. Соответствующий метод – иммуноауторадиография – был предложен Hochwald et al. (1961) (16). Метод был основан на введении в систему С14аминокислот с последующей ауторадиографией отмытых иммунофореграмм, где полоса антигена могла быть идентифицирована. Метод давал качественный анализ синтеза (включения меченой аминокислоты) в различные антигены, выявляемые на иммунофореграмме. Он был прост, красив, высоко-специфичен и не требовал предварительной очистки антигена. Нам понадобился этот подход для определения места синтеза АФП в онтогенезе и в организме опухоленосителя: в опухоли или в печени в ответ на рост опухоли идет синтез АФП? Это был принципиальный вопрос, и инкубация опухолевых клеток и клеток печени новорожденных мышей с С14глицином, приведшая к включению С14 в полосу АФП дали однозначный ответ. Далее мы отработали оптимальные условия для выявления меченого антигена в системе на немеченых носителях для качественного и полуколичественного определения степени включения метки, и попытались на основе аналитической иммунофильтрации определить абсолютную скорость синтеза АФП в многокомпонентных системах по включению метки в зону очищенного антигена (Абелев, 1965; Abelev, 1965; Абелев и Бакиров, 1968).
Таким образом, иммунодиффузия и вся система основанного на ней анализа по мере возникновения новых задач расширяла область своих возможностей. В течение двух десятилетий она оставалась определяющим методом иммунохимического анализа.
Очень серьезный вклад в систему иммунохимического анализа, главным образом на модели АФП, внесли работы А.И. Гусева и А.К. Язовой, наладивших и усовершенствовавших аналитический и препаративный электрофорез в полиакриламидном геле (ПАГ). А.И.Гусев сконструировал и сделал сам прибор для препаративного электрофореза в ПАГ (Гусев, 1969) и впервые в миллиграммовых количествах выделил АФП человека (Гусев и Язова, 1970 а). Далее они ввели в обиход лаборатории иммунизацию кроликов в лимфоузел (Гусев и Язова, 1970 б) и получили сильнейшие антисыворотки к АФП. Созданные на этой основе тест-системы на АФП человека стали основой для всех последующих исследований на человеческом АФП и для производства соответствующего диагностического набора.
Таким образом, иммунодиффузия позволила работать с индивидуальными антигенами в сложнейших системах, идентифицировать их, выявлять с высочайшей чувствительностью, выделять, сравнивать друг с другом, локализовать на срезах и изучать их биосинтез. Я надеюсь, что мы смогли создать систему, позволяющую полностью реализовать возможности иммунодиффузионного анализа применительно к изучению тканевых и опухолевых антигенов. Ch.Friend в своей рецензии на английское издание книги Zilber & Abelev «The Virology and Immunology of Cancer», 1968, писала, что каждый, кто работает с иммунодиффузией, должен познакомиться с этой книгой.
Высокочувствительные иммунодиффузионные методы
Революцию в иммунохимическом анализе вызвал радиоиммунный анализ (РИА), а затем ELISA и – еще более радикальную – введение моноклональных антител (МкАТ). Первые – благодаря очень высокой чувствительности, в 1000 раз превышающей иммунодиффузию, количественности и возможности серийных автоматических определений. Вторые – из-за абсолютной специфичности, высочайшей разрешающей способности и технологичности. Оба подхода – оправданно и неоправданно – стали вытеснять иммунодиффузию с конца 70-х и начала 80-х годов. Мы не могли смириться с этим и сконцентрировались вначале на том, чтобы довести чувствительность иммунодиффузии до РИА или ELISA. Я невзлюбил оба этих метода, прежде всего, за то, что они были лишены внутренней разрешающей способности, т.е. требовали предварительно высокоочищенных антигенов или антител и были применимы только к ним, а также за то, что они были обставлены контролями, учитывающими фон, неизвестно откуда берущийся, и, в отличие от иммунодиффузии, были сугубо эмпирическими. Несмотря на мои субъективные вкусы оба метода были налажены и используются в лаборатории (Андреев и Григорьева, 1996; 1998; 2001; Язова и др., 1990). Мы же старались резко повысить чувствительность иммунодиффузии, сохраняя все ее преимущества.
Первым шагом, о котором я уже писал, было усиление преципитатов солями Cd++ и оно примерно вчетверо увеличивало чувствительность определений, позволяя работать с более разведенными тест-системами. Однако, при этом все наименее стабильные белки выпадали в осадок и мутные ореолы появлялись вокруг лунок с антигенами и антисыворотками, мешая анализу результатов.
Вторым шагом стало выявление невидимых преципитатов I125-меченными антителами к IgG, что было предложено английским иммунохимиком D. Rowe в 1969 г. Этот способ в сочетании с предельным разведением тест-систем, дающим максимальную чувствительность, позволял определять антиген до концентрации 30–50 нг/мл (Эльгорт, Абелев, 1971; Эльгорт, 1973; Abelev et al. 1979). Чувствительность метода в точности соответствовала порогу, выше которого начинались патологические уровни АФП в сыворотке крови, и метод оказался вполне адекватным для высокочувствительного определения АФП при гепатомах и тератобластомах, при патологиях печени и беременности, не теряя абсолютной специфичности. Он был пригоден для анализа десятков или 100-200 патологических сывороток (см. раздел «Альфа-фетопротеин в иммунодиагностике»), но был слишком громоздок при переходе ко многим сотням и тысячам определений, необходимых, например, для мониторинга беременности. Мы убедились в этом на собственном опыте (Д.А. Эльгорт, 1973; А.А. Соколенко, 1980) при попытке эпидемиологического исследования, что оказалось нереальным. Дальнейшее повышение чувствительности на основе разведенных тест-систем упиралось в свойства агарового геля – способность удерживать комплекс антиген-антитело в решетке геля. Этим определялся абсолютный предел чувствительности метода. Чтобы преодолеть его и выйти на чувствительность РИА или ELISA с сохранением разрешающей способности иммунодиффузии, мы перешли на полиакриламидный гель (ПАГ), обладающий меньшим размером пор и позволяющий предварительную концентрацию антигена при электрофорезе в неоднородной системе буферов. Мы разработали модификацию иммунодиффузии, в которой антиген вначале концентрировался в плоской пластине ПАГ электрофорезом в неоднородной буферной системе, а затем идентифицировался с помощью высокоразведенной тест- системы [ Рис. 8 ].
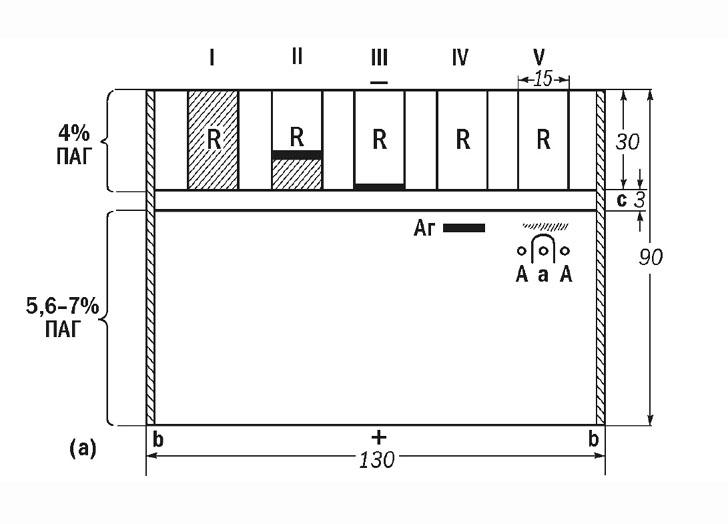
Рис. 8. Схема электрофорез-преципитации в полиакриламидном геле (ЭПАГ). R - резервуар для антигена; А - антиген-тест-система; а - антисыворотка тест- системы; I-V - последовательные стадии реакции; I-III - концентрация антигена (Аг); IV-V - включение антигена в пластину ПАГ; V - выявление антигена иммунодиффузией с тест-системой. Размеры указаны в миллиметрах. (Абелев и др., 1979) |
Образующиеся в невидимой зоне полосы преципитации выявлялись I125–анти-IgG. Метод получил название электрофорез- преципитации в полиакриламидном геле (ЭПАГ). Чувствительность вышла на уровень РИА (~ 1 нг/мл) при сохранении специфичности иммунодиффузии (Абелев, 1973; Abelev et al., 1976; 1979).
Метод позволил нам с легкостью обнаружить фоновые уровни АФП в нормальных сыворотках взрослых мышей. Однако, он был весьма громоздким (ПАГ-электрофорез, иммунодиффузия в ПАГ, обработка геля I125-анти-IgG, ауторадиография) и не количественным, т.к. не позволял определять концентрацию антигена по непрерывной шкале. Кроме того, он требовал довольно больших объемов пробы, из которых происходила концентрация антигена. Мы и сами понимали, что ЭПАГ при серийных определениях антигена может служить, главным образом, для проверки специфичности РИА в сомнительных случаях. У нас же в это время стояла задача определения продукции АФП одиночными клетками, т.е. определения минимальных количеств антигена в минимальных объемах. Для этих целей мы модифицировали ЭПАГ и разработали ультрамикрометод количественного определения антигенов (Микро-ЭПАГ), в котором проба в несколько мкл вносилась в капилляр в смеси с концентрирующим ПАГ. Батарея капилляров заливалась в плоскую пластинку ПАГ, концентрировалась электрофорезом в неоднородной системе буферов и концентрат, выходя из капилляра, включался в пластину ПАГ, смешанную с антисывороткой к изучаемому антигену. После диффузии в антитела (по Мансини) образовывалось кольцо преципитата, площадь которого была пропорциональна количеству антигена в пробе. Это кольцо выявлялось I125-анти- IgG [ Рис. 9 ] (Абелев и Перова, 1975; Abelev et. al, 1976; 1979).
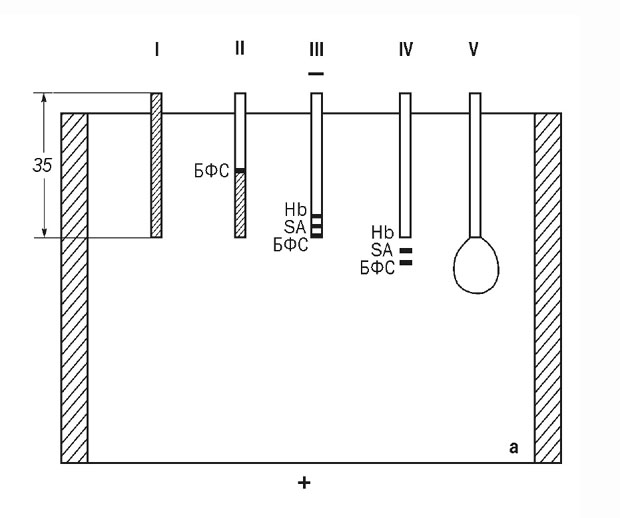
Рис. 9. Схема микроварианта электрофорез-преципитации в полиакриламидном геле (микроЭПАГ). Препарат вносится в капилляры в смеси с концентрирующим гелем. I-III - концентрация препарата; IV - включение в гель с антисывороткой; V - иммунодиффузия с образованием ринга преципитации; гемоглобин (Hb) и бромфеноловый синий (БФС) - свидетели концентрации (Абелев и Перова, 1975) |
Метод позволил определять продукцию АФП в микрокаплях, в которых инкубировались одиночные клетки или микроколонии гепатомы (Abelev, 1976; Перова и др., 1977). Он не был очень сложен, и мы даже пользовались им вместо РИА для определения раковоэмбрионального антигена (РЭА) в сыворотках больных (Карамова и др. 1976). И хотя мы изготовили довольно простое оборудование для серийных определений РЭА, метод все-таки не мог в последующем конкурировать с гораздо более технологичными РИА и ELISA, для проведения которых стали выпускать стандартные наборы (киты).
Продукция антигенов одиночными клетками. Обратный локальный гемолиз
Микро-ЭПАГ позволял в принципе определять продукцию антигена одиночными клетками, но это был «штучный» анализ – исследование микрокапель, в которых инкубировались одиночные клетки. Кроме того, нашей задачей было исследование этой продукции в динамике с тем, чтобы охарактеризовать по этому признаку клетки гепатомы и определить, каково будет их потомство по способности к продукции АФП. Микро-ЭПАГ в принципе подходил для этих целей, но позволял работать только с небольшим числом клеток, поэтому мы воспользовались совсем другим подходом, в основе которого лежал так называемый «обратный» локальный гемолиз в геле, существенно модифицированный нами для динамического исследования судьбы одиночных опухолевых клеток. Суть метода сводилась к тому, что суспензия одиночных клеток смешивалась в геле агарозы с суспензией эритроцитов, поверхность которых была конъюгирована с антителами к испытуемому антигену. Смешивание проводилось в расплавленном растворе агарозы, которая после застывания образовывала гель с взвешенными в нем клетками. После короткой, 3-х часовой, инкубации в систему добавлялись антитела к антигену, в нашем случае к АФП, и комплемент. Вокруг клеток, секретировавших АФП, эритроциты связывали АФП, затем анти-АФП, и комплемент, который вызывал их лизис, образуя видимый плак. В центре плака находилась секретирующая антиген клетка. Плак отмечался с обратной стороны пластиковой чашки. После аккуратного отмывания комплемента опухолевая клетка продолжала делиться, а ее потомство образовывало клон. Клоны фиксировались и после удаления геля окрашивались иммуногистохимически антителами к АФП. Таким образом идентифицировались АФП- продуцирующие и АФП-непродуцирующие клетки, определялась их клонообразующая способность и характеризовались клетки клонов по их продукции АФП. При этом анализ производился одновременно на десятках клеток (Эрайзер, 1977; Эрайзер и Абелев, 1982; Eraizer & Khamzina, 1988; Эрайзер и др. 1977; 1979; 1987). Это был довольно простой и очень своеобразный подход, решавший задачу, к которой мы подступали много лет – идентифицировать стволовые клетки в опухоли и определить их отношение к АФП-продуцирующим клеткам. Другими словами, выяснить, не являются ли АФП-продуцирующие клетки терминально дифференцированными, утратившими способность к пролиферации.
Иммунодиффузия и изотахофорез на ацетат-целлюлозе
В методическом отношении «обратный гемолиз» был отходом от системы иммунодиффузионного анализа, изложение которого я остановил на микро-ЭПАГ'е. Оба варианта иммунодиффузии, использующие ПАГ, затруднялись длительной отмывкой ПАГ – вначале от непрореагировавших реагентов, а затем от I125-анти-IgG. Чтобы исключить или сократить эту процедуру, мы попытались (по совету А.Е. Гурвича) заменить ПАГ ацетат- целлюлозной мембраной (АЦМ), легко и быстро отмывающейся от белков.
Еще до нас было показано, что АЦМ является хорошей подложкой для электрофореза, иммунодиффузии и иммуноэлектрофореза. Английский иммунохимик J.Kohn был в начале 70-х большим энтузиастом АЦМ. Основной сложностью работы на АЦМ было ее быстрое подсыхание и всасывание реагентов в пленку, что искажало диффузию, лежавшую в основе метода. Мы стали работать на АЦМ под слоем минерального масла, что существенно облегчило операции и исключило влияние подсыхания пленки. Под маслом мы проводили собственно иммунодиффузию и иммуноэлектрофорез (см. Abelev et al., 1979).
Далее, оказалось, что на АЦМ можно проводить концентрацию антигенов в неоднородной системе буферов – на границе Cl-/глицин- ионов, точно так же, как это делается в ПАГ-электрофорезе, в концентрирующем геле. Это открывало возможности предварительной концентрации антигена на АЦМ с последующим его выявлением иммунодиффузией с тест- системой (Абелев и Соколенко, 1977). Вообще предварительная концентрация антигена на АЦМ и использование АЦМ для иммуноэлектрофореза привело к созданию нескольких вариантов проведения иммунодиффузии и ИЭФ с повышенной чувствительностью. Среди них микровариант иммуноизотахофорез (ИТФ) на АЦМ (Абелев, 1978), «двухярусный» рокет-иммуноэлектрофорез (Соколенко и Абелев, 1979) и проведение электрофорез-преципитации, аналога ЭПАГ, в сочетании с радиоаутографией на АЦМ (Соколенко и Абелев, 1977; Abelev et al. 1979). Все эти варианты служили как бы продолжением иммунодиффузии и иммуноэлектрофореза в «невидимую область», приближающуюся по чувствительности к РИА. Я не буду останавливаться на этих методах, т.к. они явились переходными к другим вариантам, изложенным далее, и сами по себе не использовались а нашей работе.
В это время на подходе уже была эпоха моноклональных антител, которые по-своему решали проблему специфичности и чувствительности. Моноклональные антитела (МкАТ) обладают абсолютной специфичностью, т.к. продуцируются одним клоном антителообразующих В- лимфоцитов, продуцирующих антитела к одному эпитопу (17) (см. Абелев, 1996). При этом они не только высокоспецифичны, но способны выявлять «иголку в стоге сена», т.е. антигены, находящиеся в сложнейших системах в ничтожно малых количествах. МкАТ в этих случаях играет роль как бы «иммунологического микроскопа». В этом уникальная аналитическая роль МкАТ. Поскольку МкАТ направлены, как правило, к уникальным, не повторяющимся эпитопам в молекуле белков, они не создают решетку, но только небольшие растворимые комплексы. В этом случае к одной молекуле МКАТ, к каждому ее активному центру присоединяется по одной молекуле антигена, блокируя дальнейшее увеличение комплекса. Комплекс антиген-МКАТ не преципитируют, а потому не позволяют проведение иммунодиффузионного анализа. С начала 80-х годов мы начали работать с МкАТ к АФП (Язова и др., 1982; Goussev et al., 1983; Micheеl et al., 1983) и к другим антигенам (Мечетнер и др., 1983; 1985; Куприна и др., 1983). Обычно МкАТ используются в сочетании с РИА или ELISA и иммуногисто- и цитохимическим анализом. Естественно, что мы постарались связать МкАТ с иммунодиффузионными реакциями. Для сочетания иммунодиффузии с МкАТ-анализом мы разработали реакцию смешанной преципитации в геле, как бы для того, чтобы придать МкАТ преципитирующие свойства. МкАТ добавлялись к антигену, образуя растворимый комплекс, анализируемый в иммунодиффузии с поликлональной антисывороткой. МкАТ выявлялись в преципитате (Fab)1-фрагментом антител к мышиному IgG, конъюгированным с пероксидазой. Это позволяло видеть как ведут себя МкАТ в реакциях идентичности, неидентичности и частичной идентичности (Полторанина и др. 1985; Poltoranina et al. 1987). Это был хороший вариант в работе с МкАТ, но «генеральная линия» развития аналитических возможностей МкАТ в нашей работе пролегла в сочетании МкАТ с противоточным ИТФ – в методе, который мы назвали иммуноаффинной электрохроматографией (ИАЭ).
Противоточный изотахофорез. Иммуноаффинная электрохроматография
Неожиданный поворот в нашей работе наступил в 1978 г. Принципиально новые возможности открылись благодаря анализу особенностей электроосмоса в АЦМ при электрофорезе в неоднородной системе буферов, т.е. при изотахофорезе (ИТФ) (Абелев и Карамова, 1979; 1982). Сам ИТФ, основанный на электрофорезе в системе, где анодный (ведущий) буфер включал анион с высокой электрофоретической подвижностью (например Cl-), а катодный – «малоподвижный» анион (например, глицин-; или β-аланин-), как бы невозможен был теоретически в АЦМ. Ожидаемый электроосмотический отток жидкости от подвижной границы (ПГ) Cl-/глицин-, пропорциональный напряженности поля за границей, должен был быть много выше ее ожидаемого притока к границе из зоны с низким потенциалом [ Рис. 10 ].
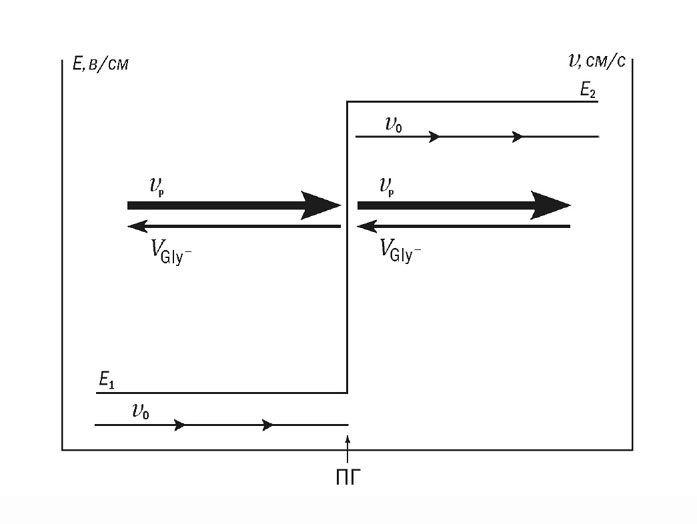
Рис. 10. Параметры системы для противоточного изотахофореза на пористой мембране: vo - ожидаемая скорость электросмотического потока, см/с; vp - результирующая скорость электроосмотического потока, Е1 и Е2 - градиент потенциала соответственно в зоне ведущего и замыкающего анионов; VCl- и Vgly- - скорость соответственно хлорид-ионов и глицина; ПГ - подвижная граница (Абелев и Карамова, 1996) |
Однако ИТФ отлично осуществлялся на АЦМ! Следовательно, пласт жидкости в АЦМ был неразрывен, а скорость электросмоса перед и за ПГ была одинаковой. Мы проверили и подтвердили эту гипотезу [ Рис. 10 ]. Из нее следовало, что ИТФ можно проводить на пористых мембранах, и что противоток жидкости перед ПГ много выше ожидаемого, а за ПГ – много ниже. Более того, гипотеза предсказывала, и это было подтверждено экспериментально, что при ИТФ на НЦМ в условиях постоянного напряжения должно наступать положение, в котором скорость миграции ПГ к аноду должна полностью уравновешиваться противотоком жидкости. Благодаря этому в АЦМ устанавливается подвижное равновесие, steady state, при котором ПГ неподвижна относительно мембраны и через нее протекает электроосмотический противоток, со скоростью, превышающей электрофоретическую миграцию любого белка. Это открывало множество новых возможностей, таких, как автоматическое проведение противоточного ИТФ, позволяющего работать с высокоразведенными растворами, благодаря одновременной концентрации и разделению белковых зон [ Рис. 11 ].
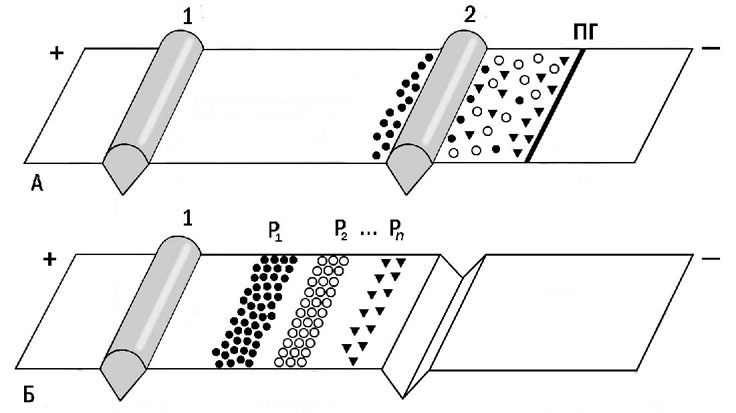
Рис. 11. Схема противоточного изотахофореза на пористой мембране (последовательные стадии концентрации (А) и разделения (Б) белков; объяснения в тексте); 1 и 2 - складки-резервуары на мембране заполнены ведущим буфером (1) и образцом (2); P1, P2, Pn - зоны разделяемых белков; ПГ - подвижная граница (Абелев и Карамова, 1996) |
Автоматика в этом методе осуществлялась без каких-либо специальных приспособлений. Стали возможны иммуноэлектрофорез и кросс-иммуноэлектрофорез, не зависящие от исходной концентрации антигенов (~ в 100 кратном интервале концентраций), автоматическое проведение многоэтапного встречного электрофореза на АЦМ, микро- и макро-препаративный ИТФ в открытой системе и иммуноаффинный электрофорез (Абелев и Карамова, 1982; 1986; Abelev, Karamova, 1984; 1987; Shranz et al. 1990). Но, пожалуй, самым важным для нас стало сочетание ИТФ на пористой мембране с использованием вместо АЦМ нитроцеллюлозной мембраны (НЦМ). АЦМ – абсолютно инертный носитель, не адсорбирующий белки. НЦМ адсорбирует белки с очень высоким сродством и может служить в качестве основы для иммуносорбента, в то время как электроосмос в ней в условиях ИТФ создает непрерывный и очень мощный противоток жидкости, как бы конвейерную ленту, переносящую любые антигены через зоны фиксированных на НЦМ антител (микроиммуносорбентов). Это дает возможность автоматического проведения многостадийных иммунохимических реакций, таких, как иммуноферментное выявление антигенов на моноклональных антителах (МкАТ) (Абелев и др. 1985 б, Abelev & Karamova, 1986; Абелев и др., 1988) или проявление иммуноблотов (Абелев, Карамова. 1988; Abelev, Karamova; 1989). И это, в свою очередь, дало самый важный для нас вариант – иммуноаффинную электрохроматографию (ИАЭ) – так называемую «зебру» (Abelev et al. 1994; Абелев и Карамова, 1996; Abelev & Karamova, 1997.). В «зебре» МкАТ к разным антигенам или разным эпитопам одного антигена фиксируются в виде поперечных полосок (слот-дотов) друг за другом на узкой полосе НЦМ. После нанесения МкАТ, НЦМ забивается иммунологически нейтральным (в данной системе) белком (казеином или яичным альбумином) и используется как носитель для противоточного ИТФ. В электроосмотический противоток вносится анализируемая система – смесь антигенов, различаемых отдельными МкАТ, а за ней проявляющая система – антитела (поли- или моноклональные), конъюгированные с пероксидазой. Антигены проходят через «зебру» из МкАТ, распределяются в соответствии со своей специфичностью и проявляются гомологичными поликлональными антителами или МкАТ к другим эпитопам, конъюгированными с ферментом [ Рис. 12 ].
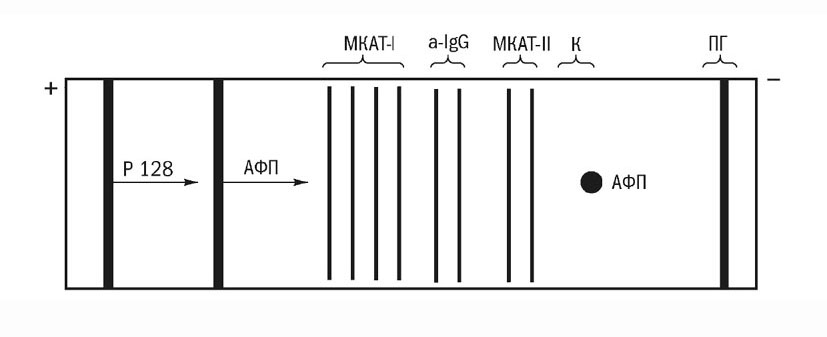
Рис. 12. Схема постановки «зебра» для выявления эпитопных вариантов альфа-фетопротеина: МкАТ-I, МкАТ-II - моноклональные антитела к разным эпитопам АФП, соответственно I и I I; a-Ig - антитела к Ig мыши; Р128 - поликлональная анти-АФП, конъюгированная с пероксидазой; К - контроль на избыток Р128 (АФП); ПГ - подвижная граница (Cl-/β--аланин-). Объяснение в тексте |
Эти этапы осуществляются автоматически, а результирующая хроматограмма проявляется в соответствующем субстрате, дающем цветную реакцию или люминисцирующий продукт. Таким образом, «зебра» позволяет разложить сложную антигенную смесь на составляющие компоненты, сравнить по специфичности разные антигены, имеющие общие или различные эпитопы, и даже разделить в микропрепаративном варианте антигены, входящие в сложную смесь. При этом весь анализ проводится автоматически, без специальной аппаратуры для автоматики и довольно быстро – в течение 3–5 часов, а его чувствительность достигает РИА. Таким образом, «зебра» на основе МкАТ, т.е. непреципитирующих антител, решает те же задачи, что иммунодиффузия, но с большей специфичностью и на уровне индивидуальных эпитопов – с предельной чувствительностью и внутренним автоматизмом. Особое значение ИАЭ приобретает в эпитопном картировании антигенов (Abelev et al. 1987; Christiansen et al. 1994; Yakimenko et al. 1998; Якименко и др. 2001) и в разделении антигена на варианты, отличающиеся по экспрессии отдельных эпитопов (Abelev et al. 1994; 2003; Karamova et al. 1998). Разделение антигенов в ИАЭ особенно эффективно сочетается с их последующим анализом в ПАГ-электрофорезе и иммуноблоттинге, – элюция вариантов при этом осуществляется кипячением слот-микросорбентов в SDS или меркаптоэтанол-SDS (Karamova et al. 1998).
Таким образом, «моноклональная революция», вытеснившая иммунодиффузию из иммунохимического анализа, в результате длительной эволюции пришла к методу, сочетающему возможности всех подходов – иммунодифузии, моноклональных антител, иммуноферментного анализа и автоматического проведения многоступенчатых реакций.
В перспективе этот метод обещает изучение белок-белковых и белок-нуклеиновых взаимодействий. Первый шаг в этом направлении уже был осуществлен (Абелев и др. 1994). Также в реальной перспективе – количественный анализ включения С14-аминокислот в разные антигены, находящиеся в одной системе.
Иммунодиагностика В-клеточных лейкозов и лимфом по белку Бенс Джонса
Противоточный изотахофорез на пористых мембранах (п-ИТФ) как будто специально был создан для определения следовых количеств моно- или олигоклональных легких цепей в моче, т.е. белка Бенс Джонса (ББД). ББД – постоянный спутник и специфический маркер плазмоцитом, где он присутствует, как правило, в очень больших количествах и может быть выявлен классическим иммуноэлектрофорезом. Можно было ожидать, что и при В-клеточных гемобластозах, где присутствуют очень малые количества моноклональных Ig, будут обнаруживаться также и очень низкие уровни ББД. Это было тем более вероятно, так как ранее было показано, что нормальные В- лимфоциты и плазмоциты продуцируют L-цепи в избытке по отношению к Н-цепям и этот избыток секретируется в среду. Кроме того была одна очень хорошая работа из Англии (18) о наличии ББД при В-клеточных лейкозах. Сразу же после разработки п- ИТФ мы проверили его пригодность для определения ББД в моче миеломного (плазмоцитомного) больного и убедились в перспективности метода. Затем мы провели систематическое исследование с Н.Е. Андреевой (кафедра гематологии ЦИУ) на В-клеточных лимфомах и получили очень хорошие результаты, которые были опубликованы у нас (Абелев и др., 1983) и доложены на международной конференции по ИТФ в Праге (Abelev & Karamova, 1984). Эта работа была специально продолжена и расширена Т.В. Синициной, аспиранткой Н.Е. Андреевой, подтвердившей на большом материале диагностическую ценность метода и его применимость для мониторинга лечения хронического лимфолейкоза (ХЛЛ) и выявления его рецидивов (Синицина и др., 1988; Андреева и др., 1989; Синицина, 1989).
Суть нашей модификации состояла в следующем: моча пациента после тщательного диализа подвергалась противоточному ИТФ на ацетат-целлюлозной мембране в присутствии амфолина, рН 3-10. Белок мочи, в том числе ББД, концентрировался и разделялся в амфолинах, после чего электрофореграмма накладывалась на агарозный гель, смешанный с поликлональной антисывороткой к Ig. Гомогенные или гетерогенные L-цепи давали характерный преципитат, по которому легко было определить их моноклональность и оценить концентрацию в моче (Abelev & Karamova, 1987; 1992).
В течение ряда последующих лет мы продолжали прослеживать больных ХЛЛ и другими В-клеточными гемобластозами, что дало вполне четкие результаты (Abelev et al., 1990; 1994). 50-60% В-клеточных лейкозов, главным образом ХЛЛ, давали ББД, динамика концентрации которых хорошо коррелировала с химиотерапией в течение ряда лет.
Таким образом, это был еще один вклад в иммунодиагностику опухолей, основанный на изучении лейкозов, и в разработку новых иммунохимических методов.
Ткане- и опухолеспецифические антигены клеточных мембран
В период работы по АФП и другим печеночным антигенам Л.А. Зильбер настаивал на том, чтобы мы переходили на работу со специфическими опухолевыми антигенами и антигенами клеточной поверхности. Но чтобы подойти к этой проблеме иммунохимически, необходимы были антитела и к клеточным поверхностям и к специфическим антигенам. Первая задача была отчасти решена получением антител к «клеточным теням», мембранной фракции, остающейся после экстракции клеточной суспензии возрастающими концентрациями NaCl от 0,14 до 1 М. Антитела к теням красили в ИФ на срезах печени и гепатом клеточную мембрану и аппарат Гольджи (Beloshapkina & Abelev, 1965). Особенно обращала на себя внимание яркая и четкая окраска желчных капилляров на границах гепатоцитов (Храмкова и Белошапкина, 1973; Рудинская и Куприна, 1975). Эта линия исследований вылилась в получение МкАТ к антигену желчных капилляров гепатоцитов, идентичному BGP1, члену семейства раково-эмбрионального антигена (РЭА) (См. «Органоспецифические антигены печени»). Эти МкАТ оказались среди наилучших в мире к этому очень важному антигену (Daniels et al., 1996).
Особое место в системе иммунохимического анализа занимают методы, направленные на выявление специфических опухолевых антигенов в сингенной системе. Первым объектом этих исследований стали мембранные антигены мышиного эритробластоза, вызываемого вирусом Раушера. Типоспецифические антитела к этому антигену были обнаружены ранее Олдом и Бойсом (Old & Boyse) в 1964 г. и мы, воспроизведя их данные, довольно успешно приступили к биохимической характеристике этого антигена (Абелев и др. 1968). Эта работа вскоре развернулась в сторону изучения группоспецифического внутреннего антигена лейкозных вирусов и выявления его в нормальных тканях как маркера эндогенных ретровирусов. Одновременно исследовалась природа типоспецифических антигенов и их отношение к собственно вирусным антигенам, почкующимся на поверхности (см. раздел «Антигены вирусных лейкозов»).
Особенно сложным было серологическое выявление специфических антигенов канцерогенных сарком, индуцированных канцерогенами. Это весьма утонченная система, где требовалась работа с первичными опухолями и опухолями самых первых генераций, контроли на полную сингенность между донором опухоли и иммунизируемой мышью по пересадке кожи, наряду с тонким ИФ анализом на живых клетках. Такая работа была выполнена Л.А. Зильбером, О.М. Лежневой и Е.С. Иевлевой, и это была первая и абсолютно точная серологическая идентификация антигенов «канцерогенных» опухолей (Лежнева и др., 1965; Lejneva et al., 1965). Но до выделения антигенов под контролем этих антител дело не дошло. Слишком нежна и трудна была эта система. Опыт же работы с ИФ на живых клетках был обобщен О.М.Лежневой (1968) и регулярно использовался в лаборатории в изучении мембранных антигенов.
* * *
Таким образом, каждый шаг в изучении антигенной структуры нормальных и опухолевых тканей был связан с разработкой адекватных новых методов, либо с модификацией имеющихся. Весь путь мы проделывали по целине и лишь при введении в область наших исследований методов собственно молекулярных первые шаги естественно делались «по протоколам» (Лазаревич, 1998). Но и здесь был предложен новый методический вариант трансфекции клеток давлением (Эрайзер и Лазаревич, 1996).
Примечания
(1) Ясно, что в этом «споре» с американской наукой, права, конечно, она, но я остаюсь при своем мнении, или, скорее, при своем вкусе. Назад
(2) Гистоны – ядерные белки, образующие прочный комплекс с ДНК – морфологическую основу хромосом. Эта работа, сделанная в самый разгар лысенковщины (в 1949–50 гг.) была опубликована лишь в 1955 г. (Белозерский и Абелев, 1955). Назад
(3) В.А. был сыном Андрея Васильевича Благовещенского, профессора биохимии Ташкентского университета, у которого учился А.Н. Белозерский. Рекомендация Белозерского была для В.А. высшим критерием, и я имел у него полные возможности для свободного выбора целей и методов работы, что было для меня в течение всей научной жизни главной ценностью. И это, несмотря на позицию препаратора, равную санитарке или посудомойке. Назад
(4) См. гл. I части 2 Назад
(5) Как рассказывал мне Г.И. Авдеев, один из наших коллег, работавший в Герценовском институте онкологии, когда он был на заседании у Министра Здравоохранения Ковригиной, где шла речь о распределении 2-х центрифуг «Spinco», Л.А. так убеждал министершу в необходимости Spinco, что заплакал (а он был железный человек). Министерша так удивилась, что дала ему центрифугу, которая около середины 60-х и появилась у нас. Назад
(6) В.А. Парнес. Вопр. патогенеза и иммунологии опухолей (п/р Г.В. Выгодчикова) М. 1956, стр. 261–263. В.А. Парнес. Иммунология лейкозов, М., 1960. Назад
(7) Институт Экспериментальной патологии и терапии рака, предшественник Онкологического научного центра. А.А. Шубин описал вирусоподобные частицы в гомогенатах опухолей человека. Назад
(8) Это была другая реализовавшаяся мечта студенческих времен. Л.А. нашел в завалах Минздрава аппарат Тизелиуса, полученный по ленд-лизу во время войны, и инженеры-оптики Красногорского оптического завода установили его у нас. Я работал на нем много лет. Электрофорез по Тизелиусу позволял получить элекрофоретические спектры сложных белковых систем, например, сыворотки крови. Этот аппарат был предшественником электрофореза на фильтровальной бумаге. Назад
(9) см. также N.G.Anderson (ed.) The development of zonal centrifuges and ancillary systems for tissue fractionation and analysis. Nat. Cancer Inst. monograph. 21, Bethesda, MD. 1966. Назад
(10) Весьма детально они рассмотрены в монографии Л.А. Зильбера и Г.И. Абелева (1962 г.) стр. 189–191. Назад
(11) Антикомплементарность антигенов – способность препарата,
содержащего антиген (экстракта, раствора гранул и т.п.), фиксировать комплемент в отсутствии антител.
Сейчас, когда о природе комплемента и его активации известно очень много, причина этого
становится понятной. С3в фиксируется ковалентной связью на частицах биологического
происхождения, не имеющих системы его инактивации. Эта система присутствует только на
клеточных мембранах млекопитающих. Назад
(12) В. Björklund & V.Björklund. Proс. Exp. Biol. Med. № 79, Р 319–323; 324–327, 1952. Назад
(13) О. Ouchterlony, Acta Path. Microbiol. Scand., № 32, 231–236, 1953. Назад
(14) Конъюгацию с арсаниловой кислотой посоветовал А.М. Оловников, тогда ст. лаб. нашей лаборатории. Назад
(15) Работу по иммунолокализации самого раково-эмбрионального антигена (СЕА) В.Н. Баранов провел в Швеции (Baranov et al., 1994). Назад
(16) Hochwald G., Thоrbecke G. & Asofsky. J. Exp.med., v. 114, 459, 1961. Назад
(17) Эпитоп – структура в молекуле антигена, распознаваемая активным центром антитела. Назад
(18) Pierson, Dartley, Stevenson & Virji, Br. J. Cancer, 41, 681–688, 1980 Назад